
UNIVERSITY OF CAPE COAST GOLDEN JUBILEE MESSAGE

SICKLE CELL AND ALLIED HAEMOGLOBINOPATHY:
THE GENETICS THAT TOUCHES YOU AND ME
Felix I D Konotey-Ahulu
ORDER OF THE VOLTA (OFFICER)
FGA MB BS MD(Lond) DSc(UCC) FRCP(Lond) FRCP(Glasg) DTMH (L’pool) FGCP FWACP FTWAS
Dr Kwegyir Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathy, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL
When The University of Cape Coast became the first in Africa to bestow a Personal Professorship and then chose me as the recipient, naming the Personal Chair after the legendary Educationalist Dr James E Kwegyir Aggrey, also known as Aggrey of Africa, UCC further honoured me by giving me the Honorary degree of Doctor of Science (Honoris Causa) on the same day that Dr Daniel Chapman Nyaho was given the Honorary Doctor of Literature Degree. As Rev Professor Samuel Adjepong, the then Vice Chancellor in the Year 2000 pronounced me “The Dr Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast, Ghana”, he commissioned me to use the honour to promote Education, Research and Service nationally and internationally in the field of “Sickle Cell Disease” in particular, and “Haemoglobinopathy” in general. The definition of these two terms follows the next paragraph, indicating how every Ghanaian family has reason to pay great attention to Haemoglobinopathy and Sickle Cell Disease.
GRATITUDE AND ORIGINALITY
I, here, record my profound gratitude for what the University of Cape Coast has done, because it did pave the way for other universities in Ghana to follow suit in creating Personal Chairs. A characteristic of my Distinguished Professorship has been that it has allowed me to latch on to one unique virtue of Dr J E Kwegyir Aggrey which was his Originality. My own definition of Originality which was the hallmark of Dr Kwegyir Aggrey is this: To think what has not been thought before, to say what has not been said before, and to do what has not been done before. It is the essence of wisdom not only to emulate people like Dr Kwegyir Aggrey [Reference 1] who were known for their originality but also to identify young people, especially students, undergraduates and postgraduates, with originality. I promptly instituted an Annual Kwegyir Aggrey Prize Examination for the Faculty of Science to do just that. That annual examination, since 2001, has been the only one in the world that allowed Candidates to take anything (books, notes, and computers) into the examination room. Why? Because the questions I set were/are meant to test originality. Answers are not found in any text book or on the internet. The 3 month assignment given to Candidates before the 3-hour paper also probes Candidates for originality. For example, take this 3-month Assignment Question which I loved to give every other year: “Describe in great detail a problem whose solution has hitherto been unsatisfactory over many years, decades, or even centuries, and suggest, also in great detail, the way you think it can be satisfactorily dealt with. This assignment tests Originality and wisdom, and I have been able to indentify UCC geniuses who have gone on to do great things after graduation. Answers to this Assignment cannot be found in any textbook or on the internet and will, hopefully be published one day. The decision to give the prizes in Guineas rather than Ghana Cedis was to keep the memory of what Gold Coast’s special gold was worth. The gold weight that sold elsewhere for 20 Shillings fetched 21 Shillings if it came from Gold Coast’s GuineaCoast. The fact of Kwegyir Aggrey’s father being not only a Linguist but also an important goldsmith at Anomabu was recalled when successful Candidates were announced on the Day of Convocation/Congregation for First Prize 100 Guineas (£105 Pounds Sterling), 75 Guineas (£78.75 Sterling) and Third Prize 50 Guineas (£52.50 Sterling). When the overall standard was high a Consolation Prize of £10 was given to each Candidate who completed the Assignment successfully (raised to £15 in 2014). The decision not to tax the University with paying these sums was also original so that a Personal Professorship need not be a burden on university finances. But the most original aspect of my Kwegyir Aggrey Distinguished Professorship was how I approached the education of the public both nationally and internationally about the hereditary ailments that constitute Sickle Cell Disease or Haemoglobinopathy which may be defined as disease caused by abnormal haemoglobins. Haemoglobin is the protein of red blood cells needed for oxygen carriage in the body. “Haemoglobin Type” is inherited through genes.
SICKLE CELL DISEASE AND HAEMOGLOBINOPATHY
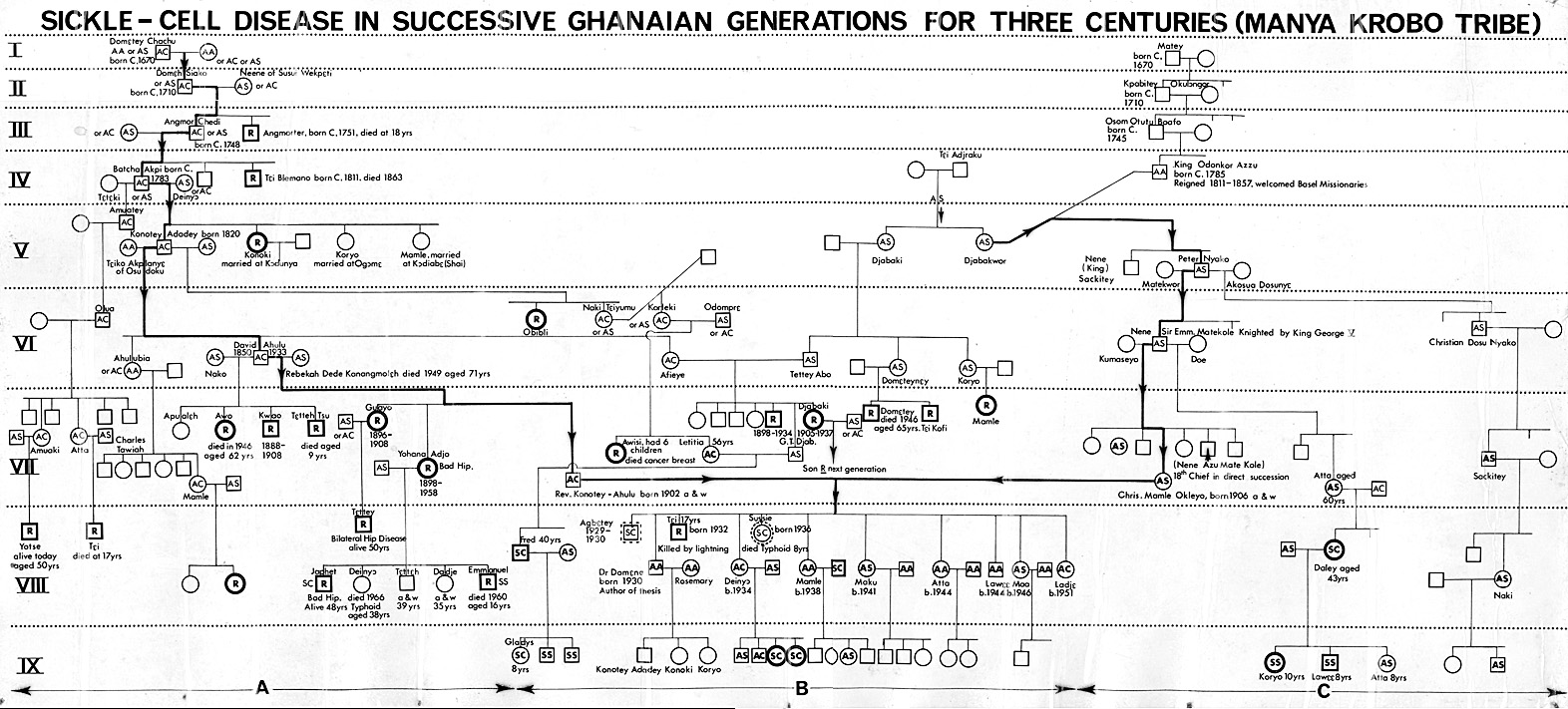
Centuries before a certain Dr James Herrick MD in Chicago, USA, in 1910 found what he called “peculiar sickle shaped red cells” in the blood of a black man who was short of blood, in other words he had anaemia, plus periodic joint pains, Ghanaians knew of a medical condition which was characterized by cold season rheumatism, yellow eyes, hip problems, leg ulcers, occasional eye symptoms, difficulty in child birth, and some other complications. Although we knew that it ran in families parents may be perfectly healthy, yet their children, some of them, would have severe joint and bone pains under certain circumstances like the rainy season, fever, violent exercise, farming, sweating a lot from very hot weather, and when suffering from infections of various kinds. Every Ghanaian tribe had a name for this, and modern experts are amazed that even when we had no microscopes to see red blood cells kinked when they should be round in shape, we in Africa knew about the condition and called it Chwechweechwe (tswetsweetswe) in Gã, Ahotutuo in Twi, Dobakotiri (Dagbani), Niwiiwii (Fante), Nuidudui (Ewe), Hemkɔm (Krobo-Dãngme), Paa (Kasena-Nankani), Koba-tuem (Buili), etc [2]. The Yorubas call it Aromolegun or Lakuregbee. The disease is so well known that my own Konotey-Ahulu family has been able to trace it generations backwards to 1670 AD [See http://www.konotey-ahulu.com/images.generation.jpg with the names of sufferers “R” for Rheumatism (also http://www.sicklecell.md/images.generation.jpg) [3] My own parents had 11 children 3 of whom were born with Hemkɔm (Sickle cell disease).
HOW IS THE DISEASE INHERITED?
How do I explain to illiterates that my father and mother did not have hemkɔm, yet 3 of us had the cold-season rheumatism? As body ache is the cardinal symptom of this illness, I use the word “ACHE” in my explanation of the inheritance of the condition. Now, every human characteristic derives its feature from contributions from both father and mother. Parents have a double code for every characteristic – skin colour, body build, type of blood protein called haemoglobin, type of Blood Group, body features, etc. Sometimes one parent’s characteristic predominates, nevertheless both characteristics are inherited. It is not always easy to say which of our characteristics is father’s, and which is mother’s but sometimes it is possible to isolate which characteristic is from father, and which from mother. The type of haemoglobin we inherit is like that. There is a code for the normal adult haemoglobin which is “A”, there is another code for a variation of normal haemoglobin known as Haemoglobin “S” which under certain circumstances like lack of oxygen stiffens up and causes the red blood cell to twist from round to sickle shape. I have given the label ‘NORM’ to normal Haemoglobin “A”, and ‘ACHE’ to any variant haemoglobin, like sickle cell haemoglobin “S”. There are many variants of normal haemoglobin, but the commonest in the world is haemoglobin “S” (for Sickle). Normal haemoglobin “A” does not cake up like “S” does with lack of oxygen. As with all other human characteristics each of us has a pair of codes (genes) for haemoglobin formation – one from father, the other from mother. The person inheriting normal haemoglobin “A” from one parent and a different haemoglobin, say “S” from the other parent is categorized “AS”, and is known as Sickle Cell Trait or “AS” phenotype [2].
Now, this is where an amazing thing happens which explains why some children of the same parents suffer from cold season rheumatism, while others do not. This is what happens: For a child to be conceived father’s sperm must unite with mother’s egg, and a father who possesses (as he must do) two hemoglobin codes and is characterized as “AS” divides his pair of codes into “A” and “S” for his millions of sperm discharged in coitus. In other words, there are sperm carrying just “A” code, and others with “S” code, never both. So you will never find a sperm with both haemoglobin codes “A” and “S” from a man who is “AS” phenotype. Is that not amazing? Similarly, a woman who is “AS” phenotype, releases her monthly egg which must carry just one of her two codes “A” or “S”, never both. Let us pause here: Dear reader, if you are not able to summarize what I have said so far about a man’s sperm, and a woman’s monthly egg, I am a poor teacher.
Now let us continue: When the man’s sperm meets the woman’s egg for fertilization 4 things can happen: (i) man’s sperm carrying “S” code meets woman’s egg carrying “A” code. The child conceived will have the phenotype “AS”, just like the parents, and will have no trouble at all with cold weather. (ii) Man’s sperm carrying “A” code meets woman’s egg with an “S” code, the child conceived will be “AS” and have no problems whatever. (iii) Man’s sperm carrying “S” code meets woman’s egg carrying “S” code conceives a child that will be “SS” who under certain circumstances will have the red cell changed into sickle shape in the blood, impeding easy flow around joints and in the bones, and causing severe pain. (iv) Man’s sperm coding haemoglobin “A” fertilizes woman’s egg coding haemoglobin “A” produces a child who will be “AA” who will not suffer cold season rheumatism. Now, the children who, like their parents, have one normal haenmoglobin “A” plus a variant haemoglobin called “S”, known as Sickle Cell Trait, cannot be distinguished except by blood test from those children who do not have any variant haemoglobin at all, “AA”. Those with haemglobin “S” from both father and mother have Sickle Cell Disease “SS”. Trait parents NORMACHE x NORMACHE produce children who may be NORMACHE (“AS” Trait – no problem), ACHENORM (“SA” Trait – no problem), ACHEACHE (“SS” sickle cell disease) and NORMNORM (no problem) [2 4] The term “Sickle Cell Disorder” is wrong because some Insurance companies use it for both Trait (no problem) and Disease to defraud.
Figure 1 Probability of AcheAche hereditary disease from NormAche Traits
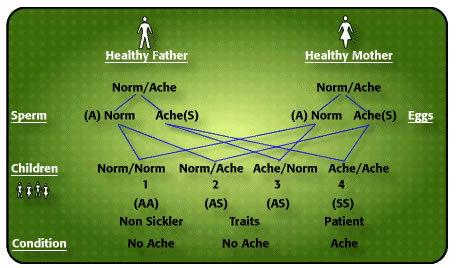
CAUTION: (i) It must never be assumed that if the first child is ACHEACHE subsequent children will not be ACHEACHE. The so-called “1 in 4” Probability of a child being born ACHEACHE is quite deceptive.
(ii) The simple test that detects whether some healthy person is carrying “S” code or gene is called the Sickle Cell Test, and can also be very deceptive. If the result is negative, in other words if there is no sickle cell haemoglobin “S” it does not mean the person does not have another variant haemoglobin which is not “S”, but which can also cause body ache when inherited with another haemoglobin variant. Please read that sentence again, and again until you understand it. The commonest such haemoglobin in Ghana is haemoglobin “C”, which is also an ACHE haemoglobin. This Haemoglobin “C” will not be detected by the Sickle Cell test. Thus my father who was sickle cell test negative was, on further investigation found to possess haemoglobin “C” that was passed on to 5 of his eleven children, 3 of whom also received the “S” code from our mother giving them two ACHE codes to form “Sickle Cell Haemoglobin C Disease”, ACHEACHE phenotype. Hence, apart from sickle cell disease “SS”, there is another haemoglobinopathy called Haemoglobin “CC” disease where the person complains of yellow eyes, tiredness, joint and abdominal pains. Sickle cell test is negative, but this is also ACHEACHE. [6 7]
HOW COMMON IS GHANAIAN NORMACHE PHENOTYPE?
Extremely common is the NORMACHE Ghanaian – 1 in every 3 healthy Ghanaian is NORMACHE “AS” or “AC”. To be precise, 20% of all southern Ghanaians are “AS” and 10% are “AC” while 20% of northern Ghanaians are “AC” and 10% are “AS”, so apart from other ACHE haemoglobins that are found in Ghana (like Haemoglobins D, F-heredirary [7 8 9], K-Woolwhich [10], Haemoglobin Korle Bu [11], Haemoglobin Osu-Christainsborg [12], beta-Thalassaemia [2], etc) 1 in 3 Ghaanaians will be found to be NORMACHE “AS” or “AC” which does them no harm whatever. This means that 1 in 9 male-female union for childbirth is that of Trait Carriers, namely NORMACHE x NORMACHE marriages, with the mother bearing ACHEACHE children. [13]
Let’s get real: : One in 3 readers of this article, 1 in 3 of the Professors at UCC, 1 in 3 members of Parliament, 1 in 3 Paramount Chiefs, 1 in 3 of the Black Star footballers, 1 in 3 market women, 1 in 3 of the Konotey-Ahulu family, 1 in 3 of all fishermen, 1 in 3 thieves, 1 in 3 prostitutes, 1 in 3 pastors, including bishops and archbishops in Ghana, 1 in 3 of the “prophets” and “faith healers”, 1 in 3 of our herbalists, is carrying an ACHE code and they probably do not know it, 1 in 3 nurses, doctors, medical students, undergraduates and post graduates are “AS” or “AC”. So are 1 in 3 rogues, and 1 in 3 liars. One in 3 of past Ghanaian Heads of State were NORMACHE. At least one of those Heads of State married an ACHEACHE lady who once consulted me. Also First Lady Mrs N M Atta Mills who was widowed not long ago told the international gathering of a thousand delegates at the International World Sickle Cell Conference, Accra in June 2010, to my astonishment, that when she was at Aburi Girls’ School I went there from Korle Bu Teaching Hospital to lecture the School on Sickle Cell Disease, then took her blood for analysis, and I later informed her in writing that she, like my own mother, had Sickle Cell Trait (“AS”)! Reader, this thing is very common paa!
GENETIC COUNSELLING AND VOLUNTARY FAMILY SIZE LIMITATION
A lot of unhelpful advice is given all around the world in connection with the possession of the ACHE gene. In Britain black pregnant women are advised to have what is called pre-natal diagnosis to see what the phenotype of the unborn baby would turn out to be. If the test reveals that the child will be ACHEACHE advice for abortion is given. I totally repudiate such advice if only because many ACHEACHE persons I know from my experience as previous Director of the largest Sickle Cell Clinic in the world have inherited from the same parents some extraordinarily brilliant genes along side the ACHE genes. Some of these ACHEACHE people are university Professors, International Judges, Senior Doctors, Diplomats, and Businesses men and women. These geniuses would have been recommended for abortion by today’s genetic counselling in Great Britain where I trained to be a doctor. A human being is far more than ACHE genes. Because of the pains they suffer when they have what is called sickle cell crises, some of these brilliant ACHEACHE persons make sure they themselves do not have ACHEACHE children. I have invented the kanad which allows them and others who are determined not to burden the next generation with ACHEACHE children to predict with 100% certainty which man-woman pairings will avoid this. This new invention [14] illustrates the kind of Originality Aggrey of Africa would have approved of. .
THE KANAD FOR PREDICTABILITY NOT PROBABILITY
Ghanaians belong to 1 of three genetic blocks when it comes to counseling with respect to what to expect when one marries whom: NORMACHE, NORMNORM, ACHEACHE. (a) A NORMNORM Ghanaian possesses no ACHE at all (b) A NORMACHE Ghanaian possesses one ACHE and one NORM code (c.) A Ghanaian possessing two ACHE haemoglobins is ACHEACHE. All 3 categories are found in my own family. There are no other categories apart from these three. My parents had abnormal haemoglobin trait so were NORMACHE. They had 11 children of whom 3 were ACHEACHE, 4 were NORMACHE, and 4 were NORMNORM. It is vitally important to know if one possesses an ACHE, and what type it is. I developed this in a pictorial form as shown below – Two groups of 3 male phenotypes, and 3 female phenotypes seeking whom to marry.

Figure 2. The kanad Male and Female Groups
I developed two groups of the three phenotypes as cubes with NORM as green, and ACHE as dark red. So NORMACHE has three sides as green, and three as dark red. ACHEACHE has all six sides of the cube as dark red, and NORMNORM has all sides of the cube green. One group of three cubes is female, and the other group of 3 is male. The six cubes are packed together into one bag to form the unit which every Ghanaian family should possess. Every Ghanaian should know which cube represents her/him. My father, for instance would be a NORMACHE green/red cube, and so would be my mother.
.
PLAYING THE KANAD
There are six possible results of Ghanaian mating when a known kanad phenotype (male) is hurled on the table together with another known kanad (female). If the parents are determined not to have an ACHEACHE child the kanad gives unequivocal results.
With relevance to sickle cell disease (scd) and other haemoglobinopathies there are 6 demonstrably different phenotype pairings of Ghanaians in procreation. Of the six pairings, as shown below, 4 produce predictable outcomes and 2 can be referred to as genetic gambling. Of the predictable outcomes one is 100 per cent disease outcome (ACHEACHE) and 3 predict 100% non-disease outcome
NB: If you carry an ACHE gene as Trait or double ACHE as hereditary disease ACHEACHE, and you do NOT want to increase the sickle cell disease burden, then go for the union that predictably leads to no births of ACHEACHE child – Respectively Number 5 and Number 3 as shown below: Marrying a NORMNORM is the only option. If, as a NORMACHE Trait, you marry an ACHEACHE person you may yet avoid having an ACHEACHE offspring, but this is gambling! If you, a trait ie NORMACHE have children with another trait NORMACHE you may produce no ACHEACHE offspring, but this is also genetic gambling which has paid off though! I have known couples then limit procreation rather than gamble further. Males need to heed this advice seriously because they have a higher MPSI (Male Procreative Superiority Index) [2 15 16 17] than females. However, if ACHEACHE children arrive, do not despair. Many are known to carry brilliant genes which their non-aching siblings do not possess. They can achieve great things in life when managed properly. (See www.sicklecell.md or www.konotey-ahulu.com) [2 18]
Predictability & Unpredictability of Producing Sickle Cell Disease (ACHEACHE) Children Using the kanad [Konotey-Ahulu Norm Ache Dice]
1. ACHEACHE x ACHEACHE [Predictable Outcome – All will be ACHEACHE]
2. ACHEACHE x NORMACHE
[Unpredictable Outcome – Some ACHEACHE, Some NORMACHE]
3. ACHEACHE x NORMNORM
[Predictable Outcome – None will be ACHEACHE, All will be NORMACHE]
4. NORMACHE x NORMACHE
[Unpredictable Outcome – Some NORMNORM, Some ACHEACHE, Some NORMACHE]
5. NORMACHE x NORMNORM
[Predictable – None will be ACHEACHE – Some will be NORMNORM, Some NORMACHE] These are healthy indistinguishable phenotypes so, please, identify Ache carriers by blood test and do genetic counselling before marriage.
6. NORMNORM x NORMNORM
[Predictable – None will be ACHEACHE – All will be NORMNORM]
TRUE STORIES
1. A Fante man, Methodist Lay Preacher, married a seamstress. They both knew they had Nwiiwii, but little did they know that however many children they had they would all be ACHEACHE. Mrs ‘D’ became pregnant 12 times, and had 13 children including twins.
This ACHEACHE x ACHEACHE couple produced an all time world record in Medical Genetics which we published in the British Medical Journal (Konotey-Ahulu and Ringelhann 1969; Bentsi-Enchill and Konotey-Ahulu 1969). The ACHEACHE of Mr ‘D’ consisted of sickle cell gene “S” and Haemoglobin “C” gene, while Mrs “D” was Sickle Cell gene “S” and “beta- Thalassaemia” for Sickle cell beta-Thalassaemia disease [19 20]
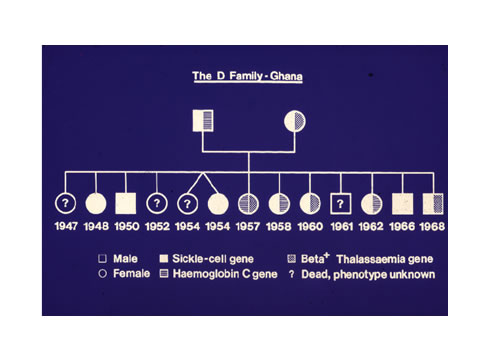
Figure 3: ACHEACHE x ACHEACHE produce 13 ACHEACHE offspring [19 20]
2. A Ghanaian Physician Specialist married a doctor, and they had 4 children with no health problems whatsoever. They thought they had “finished”, but 5 years after the last child what they called “an unplanned pregnancy” occurred and they had a lovely little girl. By the age of 12 months the child began to have signs and symptoms of what they both knew implied sickle cell disease (hand-foot syndrome). They realized they must both have been carrying sickle cell trait NORMACHE (“AS”) and never tested themselves before. Although the girl was lovely, and was indeed the most brilliant of all their children they said they found the need for Genetic Counselling and Voluntary Family Size Limitation even when couples did not suspect they carried Sickle Cell Trait.
3. This “T-S” Kwahu family knew they were both sickle cell trait NORMACHE “AS” and said they were told that 1 in 4 of any children they had would be “SS” ACHEACHE. They thought that was fair enough Genetic Gambling, and as they were tremendously in love they went ahead to produce a family. When first child, a boy, was “SS” they thought, they could have more children because the 1 in 4 had already come, and they were looking forward to the following 3 children without the ACHEACHE combination. Read the story in my book “The Sickle Cell Disease Patient”, pages 500 & 501: Second child, boy, “SS”, third “SS”, fourth “SS”, fifth “SS” and sixth “SS” [2]. But why did they continue having so many children? Answer: Because the only girl among the 6 children had died young, and Mrs T-S desperately wanted a girl. Moral of this true story is that it is wrong to tell people that Genetic Gambling will produce the different phenotypes in a certain order. As the first 4 children of the NORMACHE doctors mentioned above turned out without being ACHEACHE phenotypes so it does happen that other NORMACHE parents have been known to produce children all of whom are ACHEACHE – the term gambling will not have any relevance otherwise. The “1 in 4” prediction advice is deceptive and must be pointed out in any competent genetic counseling exercise.
4. Male Procreation needs to be curbed as part of Genetic Counselling and Voluntary Family Size Limitation (GCVFSL). On Saturday June 19 1971, Mr Kofi Sampani pulbished in The MIRROR (Accra) a front page story from Sunyani of Okomfo Klutse with a picture of his 23 wives who produced 94 children [21]. Okomfo Klutse could be NORMACHE, or NORMNORM or ACHEACHE “SC”, and no less than 8 of his wives would be carrying an ACHE gene. If he was ACHEACHE then all 94 children would have one of his ACHE genes, and any of the wives who gave an ACHE gene would have a child or children suffering from Ahotutuo. If this man was NORMACHE, like my own father was, then 47 of his children would receive an ACHE gene, and some of the children would be ACHEACHE that resulted from some of the 8 wives passing on their ACHE genes. A year earlier in 1970 I had said in the Journal of Tropical Medicine and Hygiene in England that polygamy could increase the burden of sickle cell disease [22], so males with the Trait (NORMACHE) should limit family size because of their high MPSI (Male Procreative Superiority Index) a genetic index that I invented [2 15-17] to prove that geneticists must not say that Malaria/Sickle Cell Trait Balanced Polymorphism was the main reason for the high incidence of the sickle cell trait in African populations. [23 24]. Professor George Bonney and I showed mathematically the role of polygamy in Population Genetics [25]. Incidentally, there is a high incidence of sickle cell trait among white populations in Greece, Turkey, and India [13 26].
OUR DUTY FOR THE PRESENT AND FUTURE
Populations all over the world are looking for governments to solve their health problems for them. We must be careful not to extend this “Government will do it all for me” attitude to Genetic Disease. Governments can make it easier for each of us to find out whether we are NORMACHE or NORMNORM or ACHEACHE to determine what exactly does our ACHE represents: “S”?, or “C”?, or “F-hereditary”?, or “beta-Thalassaemia”? or “Haemoglobin Korle Bu”? or Haemoglobin Osu-Christiansborg?”, or “K”?, or “D”?. True, our governments can help us [27], but we need ourselves to do something about our health, and our children’s health. My Distinguished Professorship advises you thus: “DO IT YOURSELF!”
Figure 4: Service, Education, and Research Dimensions of Genetic Counselling
This SCHEMA is the way forward. Let us know that if we are NORMNORM our grand children could be ACHEACHE. How is this possible? ANSWER: Because our husband, or wife may be a trait carrier – quite harmless – your son or daughter with the Trait may marry another Trait, like my parents did, and your grand child may then become ACHEACHE. This reasoning is quite simple, so please take this Genetics on board. As I have implied earlier it is no business of Genetic Counselling to ask a NORMACHE never to marry another NORMACHE, or to tell people how many children to have, or to tell them that the chances of hereditary disease in the offspring are such and such. No, my business as a Counsellor is to tell people the facts about the illness, and in this I seek the help of those who have ACHEACHE and who have become great Achievers in various fields of endeavour despite their medical history, and to ask them to join me in advising people to find out what phenotype they belong to, and then put to them their options: (1) Go in for Genetic Gambling, that is the “Probability” route, (2) Go in for the “I have made up my mind to avoid ACHEACHE” route which can be done by using the kanad – the Konotey-Ahulu Norm Ache Dice – to predict with 100 per cent certainty which pairings will always, without fail, avoid the ACHEACHE burden. This involves some painful decisions but has to be done, especially in the UK where many ACHEACHE patients have died from being given Morphine and Diamorphine (Heroin) for sickle cell pain [28 29 30]. Ghanaians have protested about this [31-37], but the Opiods (Opiates) for pain in sickle cell crisis have been officially sanctioned for the UK [38].If you are going to bring up children in the UK, and there is the possibility that if you have more children they will take their parents’ ACHE genes then take note that when they develop pains they will be put on Heroin and Morphine. Think seriously of limiting your family size even when you are most anxious to have another child.
A warning here for Ghanaian Doctors: Please do not adopt British or American methods of managing sickle cell disease when Ghana has produced the largest number of ACHEACHE Achievers in the world [39] without using Morphine or Diamorphine or Hydroxyurea or monthlty blood transfusions as they do abroad.
EXTEND UCC’s ORIGINALITY
I heartily congratulate The University of Cape Coast at their 50th Anniversary for evidences of their Originality – thinking what has not been thought before, saying what has not been said before, and doing what has not been done before. Ayenyekoo! – The plural for “Well Done!” Please extend your Originality to advising every student, teacher, administrative staff, and their families to find out (at their own cost) what their haemoglobin phenotype is. Let this University be the First in the world to have this policy. “At their own expense” is always better because it separates the really serious from the rest. “Save some money to enable you test yourself and your present family, and potential family” must be a Motto. One of the reasons for my KÁGÈ SICKLECELL FOUNDATION (KÁGÈ is Konotey-Ahulu Genetic Epidemiology) [38] is that governments every where have a great reluctance in dealing with genetic disease (except to decree that pregnancies should be terminated), and I want a Foundation that lays emphasis on teaching and on ethical matters in prevention processes. I have been spreading the message in lectures and international medical journals, and I have a website that is consulted around the clock from around the world [14] Since the Year 2000 when the University of Cape Coast made me The Kwegyir Aggrey Distinguished Professor of Human Genetics I have by August 2014 published 172 articles [40] most of which can be Googled and read on line, and all of which include the University of Cape Coast address, emphasizing ethical practice in Human Genetics and Clinical Medicine. May this University go from strength to strength, and may we thank GOD and take courage.
ACKNOWLEDGEMENTS
University of Cape Coast: I thank Vice Chancellor Rev Professor Samuel Adjepong and University Council, and subsequent Vice Chancellors Rev Professor Emmanuel Adow Obeng, Professor Jane Naana Opoku-Agyemang, Professor D D Kuupole, Pro-Vice Chancellors Professor Haruna Yakubu and Colleagues, and Registrars Mr Kofi Ohene and Colleagues of the Vice Chancellor’s Office and the remarkably cooperative Deans of the Faculty of Science since Year 2001 Professor Kobina Yankson, Professor Victor Gadzekpo, Professor Samuel Yeboah-Mensah, Professor B K Gordor, Professor Basuah, and their extremely hardworking Faculty Officers Rev Evelyn Nickel, Mr P K Mensah, Dr Alexis Akanson, Mr Barsi-Serbeng, Mr Ebenezer Aggrey, Ms Mary Obimpeh and their Administrative Staff Ms Emma Bedzo, Mrs Marian Aidoo, Ms Ernsetina Pokua all of whom supervised the Annual Dr Kwegyir Aggrey Prize Exams most admirably. Encouragers were Professors Jeurry Blankson, Eric Anum, Lawrence Owusu-Ansah, John Micah, and Mr Daniel Essel, Mr John K Edumadze, and Mr Sitsofe Tettey.
Indebted I am to a multitude – I am most grateful to the following who were generous in their help, financial and otherwise: Adomako, Albert & Fitnat; Acquaah-Harrison, Asabea; Chapman Nyaho, Mawunu; Akufo-Addo, Edward & Mrs Irene; Owusu, Maxwell; Botchway, Dr Kwesi; Vanderpuije Nii Oti Ahmed and Mrs Risikatu; Firth Drs Peter and Jean; Chew Dr George and May; Tettey Rev Ben and Mrs Margaret; Draisey Dr Mike; Norris Dr Geoff; Djabanor Dr Frank and Mrs Amelia; Matekole Professor Mike Ossom; Archampong Professor E Q; Addy Professor Jonathan H; Asirifi Professor Yaw; Laryea Mr Reginald; Amanor-Boadu, Dorothy; Thompson, Mrs Rosemary; Bruce-Tagoe, Professor Alexander; Codjoe, Lawyer Raymond; Mould-Iddrisu, Mrs Betty; Ekem, Dr Ivy; Tsikata, Capt Retired Kojo; Allotey, Professor Francis; Djangmah, Professor Jerome; Wosornu, Professor Lade; Hamidu, General (Retired) Joshua; Senanu, Professor Kojo; Perbi, Reindorf B; Konotey-Ahulu, Dawid & Rachel; Konotey-Ahulu, Mrs Rosemary; Madubunyi, Boniface; Awoonor, Professor Kofi; Baffour Ankomah; Nene Sakite II, Konor of Manya Krobo; Odoteye Samuel; Gbedemah, Mensah and Sophie; Omaboe, Nortey; Omaboe, Mrs Letitia; Ampim, Kofi & Deborah; Djamson, Dr Eric and Mrs Gladys; Shoetan, Samuel & Cecilia; Bonney, Professor George and Mrs Efua; Dodoo, Professor Benjamin; Awwad, Mr Awwad FRCS; Tetteh, Richard; Savage, Hugh; Badger-Laryea, Ms Christiana; Gorbett, Miss Ann; Bukar, Alhaji Ibrahim; Falae, Mr Samuel; Madueke, Mr Chike; Frimpong-Boateng, Professor Kwabena; Quartey, Dr Samuel (Philadelphia); Angelou, Professor Maya; Kofi, Tetteh J; Mlapa, Joel; Whest, Mrs Adelaide; Kofi-Tsekpo, Professor Mawule; Thairu, Professors Kihumbu and Wanja; Mensah Dr Justice Thomas and Mrs Akosua; Alexandra Road Congregational Church Hemel Hempstead, Herts, England; Ghana College of Physicians and Surgeons (Rebecca Amoh, Professors Paul Nyame, David Ofori-Adjei, J T Anim, Alex Bruce-Tagoe); Ghana Academy of Arts & Sciences (Presidents F T Sai, Letitia Obeng, R F Amonoo; F K Allotey); Academy for the Developing World (TWAS – Triest, Italy, Professor Mohamed Hassan); African Academy of Sciences (Nairobi – Professor Eng. Shem Arungu-Olende).
REFERENCES:
1 Konotey-Ahulu, FID. Aggrey of Africa: “Only the best is good enough for Africa”. New African, October 2004, pages 50-51
2 Konotey-Ahulu, FID. The Sickle Cell Disease Patient. Macmillan Education Ltd London 1991/1992. Foreword by Roland B Scott, MD (HowardUniversity) – 36 chapters with 4,500 references, 643 pages. ISBN: 0-333-39239-6 [Also Tetteh-A’Domeno Company (T-A’D Co 1996) UK – P O Box 165 Watford WD17 3ZH]
3 Konotey-Ahulu FID. The Human Genome Diversity Project: Cogitations of An African Native. Politics and the Life Sciences (PLS) 1999, Vol 18: No 2, pp 317-322. [Invited Commentary on Professor David Resnik’s article: The Human Genome Diversity Project: Ethical Problems and Solutions.] PMID: 12561789
4 Konotey-Ahulu, FID. Sickle Cell Disease. The Case for Family Planning. Accra, Astab Books Ltd, 1973; 32 pages.
5 Konotey-Ahulu, FID. Survey of Sickle Cell Disease in England and Wales. British Medical Journal 1982; 284(6309): 112 doi: 10. 1136/bmj.284/6309/112a Jan 9 1982
http://www.bmj.com/cgi/reprint/284/6309/112a.pdf
6 Konotey-Ahulu FID. The spectrum of phenotypic expression of clinical haemoglobinopathy in West Africa. New Istanbul Contribution to Clinical Science 1978 Dec; 12(3-4): 246-257. PMID: 756535 [PubMed – indexed for MEDLINE]
7 Ringelhann B, Konotey-Ahulu FID, Lehmann H and Lorkin PA. A Ghanaian adult, Homozygous Persistence of Foetal Haemoglobin and Heterozygous for Elliptocytosis. Acta Haematologica (Basel) 1970; 43(2): 100-110. PMID: 4986189 [PubMed – indexed for MEDLINE] http://lib.bioinfo.pl/pmid:4986189
8 Ringelhann B, Acquaye CTA, Oldham JH, Konotey-Ahulu FID, Yawson G, Sukumaran PK, SchroederWA and Huisman THJ. Homozygotes for the hereditary persistence of fetal haemoglobin: The ratio of G to A chains and biosynthetic studies. Biochem Genet 1977 Dec; 15(11-12): 1083-1096. PMID: 603615 [PubMed – indexed for MEDLINE
9 Acquaye CTA, Oldham JH and Konotey-Ahulu FID. Blood-donor homozygous for hereditary persistence of fetal haemoglobin. Lancet 1977 April 9; 1(8015): 796-797. PMID: 66588 [PubMed – indexed for MEDLINE]
10 Ringelhann B, Konotey-Ahulu RID, TalapatraNC, Nkrumah FK, Price B and Lehmann H. Haemoglobin K Woolwich ( 2 2 132lys –> Gln) and its combination with Hb.S and C in two Ghanaian tribes. Acta Hematologica 1971; 45(4): 250-258. PMID: 4999133 [PubMed – indexed for MEDLINE] http://lib.bioinfo.pl/pmid:4999133
11 Konotey-Ahulu FID, Gallo E, Lehmann H and Ringelhann B. Haemoglobin Korle Bu (alpha2 beta2 73 Aspartic Acid –> Asparagine), showing one of the two amino acid substitutions of Haemoglobin C Harlem. J Med Genet 1968 June; 5(2): 107-111. (This is an example of intra-genic cross-over) http://lib.bioinfo.pl/pmid:5722880 | http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1468514
12 Konotey-Ahulu FID, Kinderlerer, JL Lehmann H and Ringelhann B. Haemoglobin Osu-Christiansborg. A new chain variant of Haemoglobin A (beta 52 D3 Aspartic Acid –> Asparagine) in combination with Haemoglobin S. Journal of Med Genet 1971 Sep; 8(3): 302-305. [PubMed – indexed for MEDLINE] PMCD: PMC 1469179 & PMID: 5097135 http://pubmedcentral.nih.gov/picrender.fcgi?artid=146917&blobtype=pdf
http://lib.bioinfo.pl/pmid:5097135
13 Ringelhann B, Konotey-Ahulu FID. Hemoglobinopathies and thalassemias in Mediterranean areas and West Africa: Historical and other perspectives 1910 to 1997 – A Century Review. Atti dell’Accademia dell Science di Ferrara ( Milan) 1998;74: 267-307
14 Konotey-Ahulu FID. Sickle Cell Resource website from Year 2000 -2012. http://www.sicklecell.md or http://www.konotey-ahulu.com
15 Konotey-Ahulu FID. Male procreative superiority in African populations: The fact established and quantified and Papp Z (Eds). Medical Genetics, Exerpta Medica 1977; 600-607.
16 Konotey-Ahulu FID. Male procreative superiority index (MPSI): The missing co-efficient in African anthropogenetics. BMJ Dec 20-27 1980; 281(6256): 1700-1702 doi:10.1136/bmj.281.6256.1700 http://www.bmj.com/cgi/reprint/281/6256/1700.pdf
http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1715685&blobtype=pdf
17 Konotey-Ahulu FID. The Male Procreative Superiority Index (MPSI): its relevance to genetical counseling. In FIFTY YEARS OF HUMAN GENETICS A Festschrift and liber amicorum to celebrate the life and work of GEORGE ROBERT FRASER Edited by Oliver Mayo and Carolyn Leach. Wakefield Press 2007 (www.wakefieldpress.com.au)
1 The Parade West, Kent Town, South Australia 5067
18 Konotey-Ahulu FID. The Third International Conference On The Achievements Of Sickle Cell Disease (ACHEACHE) Patients -19th July 2010, International Conference Centre, Accra. Taking part were adult Sickle Cell Disease Patients in various Professions. Chairman was Professor Kofi Awoonor, Chairman of the Council of State, Ghana. The first ever such Conference was in London at the Royal Society of Medicine, 1 Wimpole Street in 1993. The Second was in Accra in 1995.
19 Konotey-Ahulu FID and Ringelhann B. Sickle-cell anaemia, sickle-cell thalassaemia, sickle-cell haemoglobin C disease and asymptomatic haemoglobin C thalassaemia in one Ghanaian family. BMJ 1969 Mar 8; 1(5644): 607-612. doi:10.1136/bmj-1.5644/607
PMID: 5766126 [PubMed – indexed for MEDLINE] March 8 1969
http://www.bmj.com/cgi/reprint/1/5644/607.pdf | http://lib.bioinfo.pl/pmid:5766126
20 Bentsi-Enchill KK, Konotey-Ahulu FID. Thirteen children from twelve pregnancies in sickle-cell thalassaemia. http://www.bmj.com/cgi/reprint/3/5673/762.pdf BMJ Sept 27 1969; 3(5673): 762 doi:10.1136/bmj.3.5673.762 [In Medical Memoranda Sept 27 1969]
21 Sapanin Kofi. Man with 23 wives and 94 children. Okomfo Klutse. The Mirror, Accra, Saturday 19 June, 1971, No. 932, Front Page.
22 Konotey-Ahulu FID. Maintenance of high sickling rate in Africa: Role of polygamy. J Trop Med Hyg 1970 Jan; 73(1): 19-21 (38 references). PMID: 4906442
23 Allison AC. Protection afforded by sickle cell trait against subtertian malarial infection. British Medical Journal 1: 290-294.
24 Konotey-Ahulu FID. Balanced Polymorphism and Hereditary Qualitative and Quantitative Erythrocyte defects. Ghana Med J 1972; 11: 274-285. [Chapter 8 in Ref. 2]
25 Bonney GE and Konotey-Ahulu FID. Polygamy and genetic equilibrium. Nature 1977; 265: 46-47 (January 6 1977). doi:10.1038/265046a0 …n5589/pdf/265046a0.pdf PMID: 834239 http://www.nature.com/nature/journal/v265/n5589/abs/265046a0.html
26 Choremis C et al. Blood group of a Greek Community with high sickling frequency. Lancet 1957; 2: 1333-34
27 Boyo AE, Cabannes R, Conley CL, Lehmann H, Luzzatto L, Milner PF, Ringelhann B, Weatherall DJ, Barrai I, Konotey-Ahulu FID and Motulsky AG. Geneva WHO Scientific Group on Treatment of Haemoglobinopathies and Allied Disorders. (Technical Report) 1972; 509:83 pages.
28 Mason S. Enquiry shows poor care for patients with sickle cell disease. British Medical Journal 2008; 336: 1152
29 NCEPOD (National Confidential Enquiry into Patient Outcome and Death). Sickle: A Sickle Crisis? (2008) [Sebastian Lucas (Clinical Coordinator), David Mason (Clinical Coordinator), M Mason (Chief Executive), D Weyman (Researcher), Tom Treasurer (Chairman) info@incepod.org
30 Konotey-Ahulu FID. Poor care for sickle cell disease patients: This wake up call is overdue BMJ Rapid Response May 28 2008 BMJ 2008; 336: 1152 to Susan Mayor “Enquiry shows poor care for patients with sickle cell disease” on National Confidential Enquiry into Patient Outcome and Death (NCEPOD) REPORT “SICKLE: A Sickle Crisis? (2008)
http://www.bmj.com/cgi/eletters/336/7654/1152a#196224 | http://www.info@ncepod.org
31 Konotey-Ahulu FID. Current “hit and miss” care provision for sickle cell disease patients in the UK. BMJ Rapid Response 22 July 2008
http://www.bmj.com/cgi/eletters/337/jul11_2/a771#199135
32 Chapman Nyaho Mawunu. Poor care for sickle cell disease patient: “Pain won’t kill him, but Morphine could”. British Medical Journal Rapid Response June 17 2008. http://www.bmj.com//cgi/eletters/336/7654/1152-a#197301
33 Shoetan Cecilia. I lost my Sickle Cell Disease adult daughter minutes after being given Diamorphine intravenously when she could not breathe. British Medical Journal Rapid Response June 3 2008 http://www.bmj.com//cgi/eletters/336/7654/1152-a#196520
34 Blankson Emmanuel Jeurry. Sickle Cell Disease is managed, NOT treated. British Medical Journal http://www.bmj.com//cgi/eletters/336/7654/1152-a#197350 18 June 2008
35 Frimpong-Boateng Kwabena. Poor care for sickle cell disease patients: This wake up call is overdue http://www.bmj.com//cgi/eletters/336/7654/1152-a#196359 BMJ Rapid Response May 30 2008 BMJ 2008
36 Ekem Ivy. Care for sickle cell patients. British Medical Journal Rapid Response 9 June 2008 http://www.bmj.com//cgi/eletters/336/7654/1152-a#196848
37 Dankwa Akosua M. Sickle Cell patients deserve to live. Brit Medical Journal Rapid Response http://www.bmj.com//cgi/eletters/336/7654/1152-a#197350 11 July 2008
38 Konotey-Ahulu FID. Management of acute painful sickle cell episode in hospital: NICE guidance is frightening! British Medical Journal Rapid Response 7 June 2012 http://www.bmj.com//cgi/eletters/344/bmj.e4063?ti
39 Konotey-Ahulu FID. KAGE SICKLE CELL FOUNDATION [Konotey-Ahulu Genetic Epidemiology Sickle Cell Foundation] Launched 2 September 2011 Ghana College of Surgeons, Accra.[Professor Kofi Awoonor as Chairman, and Nene Sakite II, Konor of Manya Krobo as Guest of Honour]
40 Konotey-Ahulu FID. Publications year by year from 1965 to 2014 great proportion on Sickle Cell Disease http://www.konotey-ahulu.com/publications_articles.asp
Conflict of Interest: None declared except that 3 siblings had sickle cell disease and a family genealogical history to 1670 AD www.sicklecell.md/images/generation.jpg or www.konotey-ahulu.com/images/generation.jpg [Reference 3]
PORTRAITS

Kwegyir Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL, England
———————————————————————————————————-

MB BS MD(Lond) DSc(UCC) FRCP(Lond) FRCP(Glasg) FGCP FWACP FTWAS DTMH(L’pool)
ORDER OF THE VOLTA (OFFICER)
Kwegyir Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL, England
———————————————————————————————————-
ABSTRACT
Sickle Cell and Allied Haemoglobinopathy:
The Genetics that touches You and Me
Felix I D Konotey-Ahulu
Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast Ghana and Consultant Physician Genetic Counsellor 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL, England
Originality, without doubt, is a hallmark of the University of Cape Coast. I define Originality as thinking what has not been thought before, saying what has not been said before, and doing what has not been done before. Right from the beginning when Head of State Dr Kwame Nkrumah decided to create a Third University in Ghana national and international eyebrows were raised. But this 50 year’s Anniversary of what skeptics called “an experiment” has proved many wrong because UCC has many things to show that reflect sheer originality. Not least of which was the creation of the First Personal Chair on the African Continent to be linked to the name of an Educational Legend Dr James E Kwegyir Aggrey who thought things that had not been thought before, said things that had not been said before, and did things that had not been done before.
When Vice- Chancellor Rev Professor Samuel Adjepong, with the University Council made me The Dr Kwegyir Aggrey Distinguished Professor of Human Genetics in the University of Cape Coast and at the same time gave me an Honorary Doctorate in Science (when Dr Daniel Chapman Nyaho was also honoured with a Doctorate) I was greatly humbled and extremely grateful. I knew that work displaying originality was expected of me. This chapter contribution to the Golden Jubilee gives a little glimpse of what I have used the University of Cape Coast title to achieve. Quite apart from identifying UCC Science Graduates with genius through the Annual Dr Kwegyir Aggrey Prize Examinations, I have been ceaselessly engaged in skirmishes at the frontiers of Medicine especially where Ethics is made to take a back seat and commercial concerns promoted to the fore at the expense of patients’ health. A 32 year old son of a UCC Professor (Haemoglobin ‘SC’ phenotype) walked into a British Teaching Hospital on a Friday, and by the following Tuesday he was dead on a Heroin drip. A 32 year old Ghanaian lady (also Hb “SC” phenotype) walked into a London hospital, and within 18 hours she too was dead, with her mother, a nurse, looking on while Heroin was injected. I give the British Medical Journals ‘Full Marks’ for publishing not only my informed protests from the UCC platform, but also those of other Ghanaians including the Professor whose son died. You need to pay attention to what I have said here because Haemoglobinopathy affects all of us. Look up all the 40 references.
This article has also been published here on the Cape Coast website http://bit.ly/1DzHceM



{kind=link}
{kind=link}
{kind=link}
{kind=link}