Allow me on World AIDS Day to do some self-advertisement which some Akoras may well be pleased I did.
My book “WHAT IS AIDS?” obtainable from Amazon has been commended by some prominent people not only because I was the first person ever to travel through the AIDS afflicted African countries when the Pandemic burst on the world, and interviewed over 120 International Prostitutes on duty, but also published my findings in peer re-viewed medical/scientific journals on both sides of the Atlantic. Just take a look at some of the comments:
Professor (Justice) Thomas Mensah, Past Chairman of OAA-UK, in his most generous Book Review: “Experts generally find it easy to expatiate on their subjects to the comprehension of their fellow experts. But it takes a truly first-rate expert to make a highly technical subject understandable to laymen, without either trivialising the issues involved or sounding condescending in the process. When the technical subject in question happens to be emotive and encrusted with irrational fears and deeply ingrained prejudices, as is the case with AIDS, then it requires not just deep knowledge but also exemplary courage to undertake the task of discussing and explaining it to the uninitiated. And to be able to do this successfully calls for unique literary skills which one does not normally expect from practising scientists.
“Fortunately for the reader, Dr Konotey-Ahulu brings to the task of writing about Acquired Immune Deficiency Syndrome (AIDS) an unusual combination of professional expertise, rare courage and integrity and supreme gift of language and style. …
“Like the true scientist that he is, Dr Konotey-Ahulu would be the last to claim that his is the final word on the subject or that the prescriptions he suggests for prevention will be either successful or even feasible. What cannot be denied is that he has set the problem of AIDS in a totally different context by giving the individual man and woman, as well as African Governments and the world community in general, the information and perspectives on which to formulate a strategy for the future. For this he deserves the gratitude of all of us. Thomas Mensah”.
Professor Maya Angelou (USA): “Compulsive reading. I could not put it down.”
Mr J G Amamoo (London): “Scholarly. A remarkable book”.
Dr Bill Turner (UK): “Excellent! A must for every medical student”.
Dr Fred Wurapa (Congo): “The story of AIDS in Africa as it should be told”.
Mr Louis Obosi (Nigeria): “Written for Africans, but should find its way to U.N.O.”
Professor George Fraser (Oxford): “Excellent book … the most important topic in Medicine currently”.
Group Captain John Weir (London): “Remarkable book. Easily understood.”
Mr K Gyang-Apenteng (London): “The most commendable aspect is its sheer readability.”
Professor Geraint James and Professor Dame Sheila Sherlock (London): “Superb in every way.”
Dr Nam Dunbar (Oxford): “Clearly dispelled many false notions about the causes and spread of AIDS”.
Marion McTair (London): “ …a masterpiece of good descriptive writing”.
Dr Ann Rudwick (London): “… a tour de force”.
All the above is what others have said about my book. What do I myself say about it?
ANSWER:
(a) It is experiential in much of the discourse. What the sex ladies on duty told me is recounted in detail, forcing me to start the book with (Chapter One) TAFRACHER, that unique Ghanaian de-vulgarising prefix-word that I introduced into medical vocabulary in the British Medical Journal in 1975.
(b) Photographs of me examining the sex ladies in their homes after their business demonstrating, with their permission, what AIDS looks like.
(c) Disclosing names of self-confessed scientific liars or lying scientists (Can you identify the 5 names?)
(d) Akoras doing research will find a detailed list of references both of truth and the self con-fessed untruths to include in their work.
(e) The book contains such unusual “stuff” that a lady from “USA TODAY” rang me in London at the Cromwell Hospital to ask “Who funded you to go to African countries to research AIDS?”. When I told her “I went to my bank in South Kensington, London, to borrow money for the 6-week tour to see what is happening at the grass roots on my Continent” she appeared less interested and hung up.
[Rapid Response to Perthes’ disease by Peter Kannu & Andrew Howard
(Published 23 Sept 2014) British Medical Journal – Practice Easily Missed?]
The instructive article [1] of Peter Kannu and Andrew Howard (Oct 4, p 32) has presenting clinical
features that can equally apply to children with sickle cell disease whose omission in differential
diagnosis does not help busy Residents and House Physicians in hospitals anywhere in the world.
RADIOLOGICAL DIFFERENTIATION: PERTHES’ VERSUS SICKLE CELL DISEASE HIP
While clinical presentation – the limp with the physical signs elicited – is identical in Perthes’
and sickle cell disease hip in children the radiological features help in differentiation [2 page
239]. The latter hip differentiation “from Calve-Legg- Perthes’ disease is not difficult” [3]
especially when “the significant differences in age of onset, degree and site of epiphyseal
involvement and changes in the femoral metaphyses are considered” [4 5]. Like in Perthes’ there was
no hip involvement in 157 consecutive children with sickle cell disease aged 4 and below [2 page
239], but between 5 to 9 years of age there was 1 case in 201 consecutive sickle cell disease
patients (0.5%) and between 10 to 14 years we had 7 (4%) of 177 consecutive patients [2]. Therefore
below the age of 15 years we in Ghana found 8 cases of avascular/aseptic femoral head necrosis in
200 consecutive sickle cell disease patients (2.5%) observed by one clinician (myself). The article
of Kannu and Howard [1] states: “The annual incidence of Perthes’ disease among children under 15
ranges from 0.2 to 19.1 per 100,000” which, while quoting the paper of Perry and colleagues on the
“racial and geographic factors on the incidence of Legg-Calve-Perthes disease” [6], failed to
comment that these so-called “racial factors” could be haemoglobinopathic.
DEFINITION OF SICKLE CELL DISEASE
Sickle cell disease is defined as possession of two beta-globin gene defects at least one of which
is the sickle cell gene [2], therefore the phenotypes constituting sickle cell disease are ‘SS’
known as sickle cell anaemia, ‘SC’ known as sickle cell haemoglobin C disease, ‘Sβ-Thal’ which is
sickle cell beta-Thalassaemia, ‘SF-HEREDITARY’ for sickle cell Hereditary Persistence of Foetal
Haemoglobin which 4 phenotypes comprised the commonest sickle cell diseases seen in Ghana. More
‘SC’ phenotype (6.6% of 603 consecutive patients of all
ages) than ‘SS’ (2’8 % of 600) suffered hip necrosis, but no disease phenotype is exempt [2]
ANYWHERE IN THE WORLD
Sickle cell disease occurs in significant measure in White people in Greece, Turkey, Sicily, and in
the Middle East, India, and in their descendants in the diaspora [2, pages 76-82]. Canada where
this article originates [1] has a good representation of these Mediterranean populations [7] as
well as Blacks and though the authors, as part of their investigations carried out for Perthes’
disease mention “a full blood count” there is need to remind Residents and House Physicians
anywhere in the world that haemoglobin electrophoresis must always be counted among the tests to be
carried out in any tentative clinical diagnosis of Perthes’ disease.
FORGETTING PAST PUBLICATIONS?
One Editor of a prestigious international medical publication once wrote asking me to limit my
references to articles published after the year 2000 AD. I wrote back to point out that clinical
signs were/are clinical signs and doctors needed reminding of their significance in any generation.
As part of their treatment regime Peter Kannu and Andrew Howard list “encouraging swimming” among
exercises to be recommended in Perthes’ disease. Well, fifty years ago bar 4 months I named in
Lancet swimming among the 17 commonest precipitating causes of sickle cell crisis [8]. Mistake
sickle cell disease for Perthes’ disease and this advice to encourage swimming would be misplaced.
Fortunately, Obisesan and Bohrer [5] knew how to diagnose Perthes’ disease in Black children in
West Africa 42 years ago. Should the present authors have mentioned this? Or were they dismissing
anything published before AD 2000 as being of little significance? Just two out of their 13
references are before the 21st Century. The increasing habit of starting articles with a clinical
dimension thus: “We did a MEDSEARCH going back 25 years” is neither academic nor helpful, because
quite significant clinical material may be missed. In 1972 I discovered and described in The Lancet
a new physical sign in sickle cell disease, to the extent of quantifying its incidence in the
different sickle cell disease phenotypes, the peripheral mental nerve neuropathy of sickle cell
crisis [9] – what came to be known on ward rounds as the “kanumblll sign”. Twenty-five years later
Sally Davies and L Oni published in the BMJ a comprehensive article on Sickle Cell Disease [10]
limiting their references to cover the previous 25 years, thereby (but not deliberately) omitting a
vital physical sign (numb lower lip) that could have drawn the attention of any doctor or nurse to
the fact that the person they were confronted with had sickle cell disease.
Competing interests: None declared
Felix I D Konotey-Ahulu MD(Lond) FRCP(Lond) DTMH(L’pool) Kwqegyir
Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant
Physician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathies, 9 Harley Street Ltd,
Phoenix Hospital Group, London W1G 9AL (E-mail felix@konotey-ahulu.com Web: www.sicklecell.md)
1 Kannu P, Howard A. Perthes’ disease. BMJ 2014; 349:g5584 (doi:1136/bmj.g5584)
2 Konotey-Ahulu FID. The Sickle Cell Disease Patient: Natural History from a
Clinico-epidemiological study of the first 1550 patients of Korle Bu Hospital Sickle Cell Clinic.
The Macmillan Press Ltd, London 1991 & 1992 and T-A’D Co Watford, 1996.
3 Golding JSR. The bone changes in sickle cell anaemia. Ann Roy Col Surg Eng 1956; 19: 296-315.
4 Barton CJ, Cockshott WP. Bone changes in Haemoglobin SC disease. Am J Roentgen Rad Ther Nuc Med
1962; 88: 523-532.
5 Obisesan AA, Bohrer SP. Perthes’ disease in Nigerians. Ghana Medical Journal 1972; 11: 298-302.
6 Perry DC, Machin DM, Pope D, Bruce CE, Dangerfield P, Platt MJ, et al. Racial and geographic
factors in incidence of Legg-Calve-Perthes’ disease: a systematic review. Am J Epidemiol 2012; 175:
159-166.
7 Ringelhann B, Konotey-Ahulu FID. Hemoglobinopathies & Thalassemias in Mediterranean areas and in
West Africa: historical and other perspectives 1910- 1997 – A Century Review. Atti del’Accademia
della Science di Ferrara 1998; 74: 267-307.
Dr Kwegyir Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathy, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL
When The University of Cape Coast became the first in Africa to bestow a Personal Professorship and then chose me as the recipient, naming the Personal Chair after the legendary Educationalist Dr James E Kwegyir Aggrey, also known as Aggrey of Africa, UCC further honoured me by giving me the Honorary degree of Doctor of Science (Honoris Causa) on the same day that Dr Daniel Chapman Nyaho was given the Honorary Doctor of Literature Degree. As Rev Professor Samuel Adjepong, the then Vice Chancellor in the Year 2000 pronounced me “The Dr Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast, Ghana”, he commissioned me to use the honour to promote Education, Research and Service nationally and internationally in the field of “Sickle Cell Disease” in particular, and “Haemoglobinopathy” in general. The definition of these two terms follows the next paragraph, indicating how every Ghanaian family has reason to pay great attention to Haemoglobinopathy and Sickle Cell Disease.
GRATITUDE AND ORIGINALITY
I, here, record my profound gratitude for what the University of Cape Coast has done, because it did pave the way for other universities in Ghana to follow suit in creating Personal Chairs. A characteristic of my Distinguished Professorship has been that it has allowed me to latch on to one unique virtue of Dr J E Kwegyir Aggrey which was his Originality. My own definition of Originality which was the hallmark of Dr Kwegyir Aggrey is this: To think what has not been thought before, to say what has not been said before, and to do what has not been done before. It is the essence of wisdom not only to emulate people like Dr Kwegyir Aggrey [Reference 1] who were known for their originality but also to identify young people, especially students, undergraduates and postgraduates, with originality. I promptly instituted an Annual Kwegyir Aggrey Prize Examination for the Faculty of Science to do just that. That annual examination, since 2001, has been the only one in the world that allowed Candidates to take anything (books, notes, and computers) into the examination room. Why? Because the questions I set were/are meant to test originality. Answers are not found in any text book or on the internet. The 3 month assignment given to Candidates before the 3-hour paper also probes Candidates for originality. For example, take this 3-month Assignment Question which I loved to give every other year: “Describe in great detail a problem whose solution has hitherto been unsatisfactory over many years, decades, or even centuries, and suggest, also in great detail, the way you think it can be satisfactorily dealt with. This assignment tests Originality and wisdom, and I have been able to indentify UCC geniuses who have gone on to do great things after graduation. Answers to this Assignment cannot be found in any textbook or on the internet and will, hopefully be published one day. The decision to give the prizes in Guineas rather than Ghana Cedis was to keep the memory of what Gold Coast’s special gold was worth. The gold weight that sold elsewhere for 20 Shillings fetched 21 Shillings if it came from Gold Coast’s GuineaCoast. The fact of Kwegyir Aggrey’s father being not only a Linguist but also an important goldsmith at Anomabu was recalled when successful Candidates were announced on the Day of Convocation/Congregation for First Prize 100 Guineas (£105 Pounds Sterling), 75 Guineas (£78.75 Sterling) and Third Prize 50 Guineas (£52.50 Sterling). When the overall standard was high a Consolation Prize of £10 was given to each Candidate who completed the Assignment successfully (raised to £15 in 2014). The decision not to tax the University with paying these sums was also original so that a Personal Professorship need not be a burden on university finances. But the most original aspect of my Kwegyir Aggrey Distinguished Professorship was how I approached the education of the public both nationally and internationally about the hereditary ailments that constitute Sickle Cell Disease or Haemoglobinopathy which may be defined as disease caused by abnormal haemoglobins. Haemoglobin is the protein of red blood cells needed for oxygen carriage in the body. “Haemoglobin Type” is inherited through genes.
SICKLE CELL DISEASE AND HAEMOGLOBINOPATHY
Centuries before a certain Dr James Herrick MD in Chicago, USA, in 1910 found what he called “peculiar sickle shaped red cells” in the blood of a black man who was short of blood, in other words he had anaemia, plus periodic joint pains, Ghanaians knew of a medical condition which was characterized by cold season rheumatism, yellow eyes, hip problems, leg ulcers, occasional eye symptoms, difficulty in child birth, and some other complications. Although we knew that it ran in families parents may be perfectly healthy, yet their children, some of them, would have severe joint and bone pains under certain circumstances like the rainy season, fever, violent exercise, farming, sweating a lot from very hot weather, and when suffering from infections of various kinds. Every Ghanaian tribe had a name for this, and modern experts are amazed that even when we had no microscopes to see red blood cells kinked when they should be round in shape, we in Africa knew about the condition and called it Chwechweechwe (tswetsweetswe) in Gã, Ahotutuo in Twi, Dobakotiri (Dagbani), Niwiiwii (Fante), Nuidudui (Ewe), Hemkɔm (Krobo-Dãngme), Paa (Kasena-Nankani), Koba-tuem (Buili), etc [2]. The Yorubas call it Aromolegun or Lakuregbee. The disease is so well known that my own Konotey-Ahulu family has been able to trace it generations backwards to 1670 AD [See http://www.konotey-ahulu.com/images.generation.jpg with the names of sufferers “R” for Rheumatism (also http://www.sicklecell.md/images.generation.jpg) [3] My own parents had 11 children 3 of whom were born with Hemkɔm (Sickle cell disease).
HOW IS THE DISEASE INHERITED?
How do I explain to illiterates that my father and mother did not have hemkɔm, yet 3 of us had the cold-season rheumatism? As body ache is the cardinal symptom of this illness, I use the word “ACHE” in my explanation of the inheritance of the condition. Now, every human characteristic derives its feature from contributions from both father and mother. Parents have a double code for every characteristic – skin colour, body build, type of blood protein called haemoglobin, type of Blood Group, body features, etc. Sometimes one parent’s characteristic predominates, nevertheless both characteristics are inherited. It is not always easy to say which of our characteristics is father’s, and which is mother’s but sometimes it is possible to isolate which characteristic is from father, and which from mother. The type of haemoglobin we inherit is like that. There is a code for the normal adult haemoglobin which is “A”, there is another code for a variation of normal haemoglobin known as Haemoglobin “S” which under certain circumstances like lack of oxygen stiffens up and causes the red blood cell to twist from round to sickle shape. I have given the label ‘NORM’ to normal Haemoglobin “A”, and ‘ACHE’ to any variant haemoglobin, like sickle cell haemoglobin “S”. There are many variants of normal haemoglobin, but the commonest in the world is haemoglobin “S” (for Sickle). Normal haemoglobin “A” does not cake up like “S” does with lack of oxygen. As with all other human characteristics each of us has a pair of codes (genes) for haemoglobin formation – one from father, the other from mother. The person inheriting normal haemoglobin “A” from one parent and a different haemoglobin, say “S” from the other parent is categorized “AS”, and is known as Sickle Cell Trait or “AS” phenotype [2].
Now, this is where an amazing thing happens which explains why some children of the same parents suffer from cold season rheumatism, while others do not. This is what happens: For a child to be conceived father’s sperm must unite with mother’s egg, and a father who possesses (as he must do) two hemoglobin codes and is characterized as “AS” divides his pair of codes into “A” and “S” for his millions of sperm discharged in coitus. In other words, there are sperm carrying just “A” code, and others with “S” code, never both. So you will never find a sperm with both haemoglobin codes “A” and “S” from a man who is “AS” phenotype. Is that not amazing? Similarly, a woman who is “AS” phenotype, releases her monthly egg which must carry just one of her two codes “A” or “S”, never both. Let us pause here: Dear reader, if you are not able to summarize what I have said so far about a man’s sperm, and a woman’s monthly egg, I am a poor teacher.
Now let us continue: When the man’s sperm meets the woman’s egg for fertilization 4 things can happen: (i) man’s sperm carrying “S” code meets woman’s egg carrying “A” code. The child conceived will have the phenotype “AS”, just like the parents, and will have no trouble at all with cold weather. (ii) Man’s sperm carrying “A” code meets woman’s egg with an “S” code, the child conceived will be “AS” and have no problems whatever. (iii) Man’s sperm carrying “S” code meets woman’s egg carrying “S” code conceives a child that will be “SS” who under certain circumstances will have the red cell changed into sickle shape in the blood, impeding easy flow around joints and in the bones, and causing severe pain. (iv) Man’s sperm coding haemoglobin “A” fertilizes woman’s egg coding haemoglobin “A” produces a child who will be “AA” who will not suffer cold season rheumatism. Now, the children who, like their parents, have one normal haenmoglobin “A” plus a variant haemoglobin called “S”, known as Sickle Cell Trait, cannot be distinguished except by blood test from those children who do not have any variant haemoglobin at all, “AA”. Those with haemglobin “S” from both father and mother have Sickle Cell Disease “SS”. Trait parents NORMACHE x NORMACHE produce children who may be NORMACHE (“AS” Trait – no problem), ACHENORM (“SA” Trait – no problem), ACHEACHE (“SS” sickle cell disease) and NORMNORM (no problem) [2 4] The term “Sickle Cell Disorder” is wrong because some Insurance companies use it for both Trait (no problem) and Disease to defraud.
Figure 1 Probability of AcheAche hereditary disease from NormAche Traits
CAUTION: (i) It must never be assumed that if the first child is ACHEACHE subsequent children will not be ACHEACHE. The so-called “1 in 4” Probability of a child being born ACHEACHE is quite deceptive.
(ii) The simple test that detects whether some healthy person is carrying “S” code or gene is called the Sickle Cell Test, and can also be very deceptive. If the result is negative, in other words if there is no sickle cell haemoglobin “S” it does not mean the person does not have another variant haemoglobin which is not “S”, but which can also cause body ache when inherited with another haemoglobin variant. Please read that sentence again, and again until you understand it. The commonest such haemoglobin in Ghana is haemoglobin “C”, which is also an ACHE haemoglobin. This Haemoglobin “C” will not be detected by the Sickle Cell test. Thus my father who was sickle cell test negative was, on further investigation found to possess haemoglobin “C” that was passed on to 5 of his eleven children, 3 of whom also received the “S” code from our mother giving them two ACHE codes to form “Sickle Cell Haemoglobin C Disease”, ACHEACHE phenotype. Hence, apart from sickle cell disease “SS”, there is another haemoglobinopathy called Haemoglobin “CC” disease where the person complains of yellow eyes, tiredness, joint and abdominal pains. Sickle cell test is negative, but this is also ACHEACHE. [6 7]
HOW COMMON IS GHANAIAN NORMACHE PHENOTYPE?
Extremely common is the NORMACHE Ghanaian – 1 in every 3 healthy Ghanaian is NORMACHE “AS” or “AC”. To be precise, 20% of all southern Ghanaians are “AS” and 10% are “AC” while 20% of northern Ghanaians are “AC” and 10% are “AS”, so apart from other ACHE haemoglobins that are found in Ghana (like Haemoglobins D, F-heredirary [7 8 9], K-Woolwhich [10], Haemoglobin Korle Bu [11], Haemoglobin Osu-Christainsborg [12], beta-Thalassaemia [2], etc) 1 in 3 Ghaanaians will be found to be NORMACHE “AS” or “AC” which does them no harm whatever. This means that 1 in 9 male-female union for childbirth is that of Trait Carriers, namely NORMACHE x NORMACHE marriages, with the mother bearing ACHEACHE children. [13]
Let’s get real: : One in 3 readers of this article, 1 in 3 of the Professors at UCC, 1 in 3 members of Parliament, 1 in 3 Paramount Chiefs, 1 in 3 of the Black Star footballers, 1 in 3 market women, 1 in 3 of the Konotey-Ahulu family, 1 in 3 of all fishermen, 1 in 3 thieves, 1 in 3 prostitutes, 1 in 3 pastors, including bishops and archbishops in Ghana, 1 in 3 of the “prophets” and “faith healers”, 1 in 3 of our herbalists, is carrying an ACHE code and they probably do not know it, 1 in 3 nurses, doctors, medical students, undergraduates and post graduates are “AS” or “AC”. So are 1 in 3 rogues, and 1 in 3 liars. One in 3 of past Ghanaian Heads of State were NORMACHE. At least one of those Heads of State married an ACHEACHE lady who once consulted me. Also First Lady Mrs N M Atta Mills who was widowed not long ago told the international gathering of a thousand delegates at the International World Sickle Cell Conference, Accra in June 2010, to my astonishment, that when she was at Aburi Girls’ School I went there from Korle Bu Teaching Hospital to lecture the School on Sickle Cell Disease, then took her blood for analysis, and I later informed her in writing that she, like my own mother, had Sickle Cell Trait (“AS”)! Reader, this thing is very common paa!
GENETIC COUNSELLING AND VOLUNTARY FAMILY SIZE LIMITATION
A lot of unhelpful advice is given all around the world in connection with the possession of the ACHE gene. In Britain black pregnant women are advised to have what is called pre-natal diagnosis to see what the phenotype of the unborn baby would turn out to be. If the test reveals that the child will be ACHEACHE advice for abortion is given. I totally repudiate such advice if only because many ACHEACHE persons I know from my experience as previous Director of the largest Sickle Cell Clinic in the world have inherited from the same parents some extraordinarily brilliant genes along side the ACHE genes. Some of these ACHEACHE people are university Professors, International Judges, Senior Doctors, Diplomats, and Businesses men and women. These geniuses would have been recommended for abortion by today’s genetic counselling in Great Britain where I trained to be a doctor. A human being is far more than ACHE genes. Because of the pains they suffer when they have what is called sickle cell crises, some of these brilliant ACHEACHE persons make sure they themselves do not have ACHEACHE children. I have invented the kanad which allows them and others who are determined not to burden the next generation with ACHEACHE children to predict with 100% certainty which man-woman pairings will avoid this. This new invention [14] illustrates the kind of Originality Aggrey of Africa would have approved of. .
THE KANAD FOR PREDICTABILITY NOT PROBABILITY
Ghanaians belong to 1 of three genetic blocks when it comes to counseling with respect to what to expect when one marries whom: NORMACHE, NORMNORM, ACHEACHE. (a) A NORMNORM Ghanaian possesses no ACHE at all (b) A NORMACHE Ghanaian possesses one ACHE and one NORM code (c.) A Ghanaian possessing two ACHE haemoglobins is ACHEACHE. All 3 categories are found in my own family. There are no other categories apart from these three. My parents had abnormal haemoglobin trait so were NORMACHE. They had 11 children of whom 3 were ACHEACHE, 4 were NORMACHE, and 4 were NORMNORM. It is vitally important to know if one possesses an ACHE, and what type it is. I developed this in a pictorial form as shown below – Two groups of 3 male phenotypes, and 3 female phenotypes seeking whom to marry.
Figure 2. The kanad Male and Female Groups
I developed two groups of the three phenotypes as cubes with NORM as green, and ACHE as dark red. So NORMACHE has three sides as green, and three as dark red. ACHEACHE has all six sides of the cube as dark red, and NORMNORM has all sides of the cube green. One group of three cubes is female, and the other group of 3 is male. The six cubes are packed together into one bag to form the unit which every Ghanaian family should possess. Every Ghanaian should know which cube represents her/him. My father, for instance would be a NORMACHE green/red cube, and so would be my mother.
.
PLAYING THE KANAD
There are six possible results of Ghanaian mating when a known kanad phenotype (male) is hurled on the table together with another known kanad (female). If the parents are determined not to have an ACHEACHE child the kanad gives unequivocal results.
With relevance to sickle cell disease (scd) and other haemoglobinopathies there are 6 demonstrably different phenotype pairings of Ghanaians in procreation. Of the six pairings, as shown below, 4 produce predictable outcomes and 2 can be referred to as genetic gambling. Of the predictable outcomes one is 100 per cent disease outcome (ACHEACHE) and 3 predict 100% non-disease outcome
NB:If you carry an ACHE gene as Trait or double ACHE as hereditary disease ACHEACHE, and you do NOT want to increase the sickle cell disease burden, then go for the union that predictably leads to no births of ACHEACHE child – Respectively Number 5 and Number 3 as shown below: Marrying a NORMNORM is the only option. If, as a NORMACHE Trait, you marry an ACHEACHE person you may yet avoid having an ACHEACHE offspring, but this is gambling! If you, a trait ie NORMACHE have children with another trait NORMACHE you may produce no ACHEACHE offspring, but this is also genetic gambling which has paid off though! I have known couples then limit procreation rather than gamble further. Males need to heed this advice seriously because they have a higher MPSI (Male Procreative Superiority Index) [2 15 16 17] than females. However, if ACHEACHE children arrive, do not despair. Many are known to carry brilliant genes which their non-aching siblings do not possess. They can achieve great things in life when managed properly. (See www.sicklecell.md or www.konotey-ahulu.com) [2 18]
Predictability & Unpredictability of Producing Sickle Cell Disease (ACHEACHE) Children Using the kanad [Konotey-Ahulu Norm Ache Dice]
1. ACHEACHE x ACHEACHE [Predictable Outcome – All will be ACHEACHE]
2. ACHEACHEx NORMACHE
[Unpredictable Outcome – Some ACHEACHE, Some NORMACHE]
3. ACHEACHE xNORMNORM
[Predictable Outcome – None will be ACHEACHE, All will be NORMACHE]
4. NORMACHE x NORMACHE
[Unpredictable Outcome – Some NORMNORM, Some ACHEACHE, Some NORMACHE]
5. NORMACHE x NORMNORM
[Predictable – None will be ACHEACHE – Some will be NORMNORM, Some NORMACHE] These are healthy indistinguishable phenotypes so, please, identify Ache carriers by blood test and do genetic counselling before marriage.
6. NORMNORMx NORMNORM
[Predictable – None will be ACHEACHE– All will be NORMNORM]
TRUE STORIES
1. A Fante man, Methodist Lay Preacher, married a seamstress. They both knew they had Nwiiwii, but little did they know that however many children they had they would all be ACHEACHE. Mrs ‘D’ became pregnant 12 times, and had 13 children including twins.
This ACHEACHE x ACHEACHE couple produced an all time world record in Medical Genetics which we published in the British Medical Journal (Konotey-Ahulu and Ringelhann 1969; Bentsi-Enchill and Konotey-Ahulu 1969). The ACHEACHE of Mr ‘D’ consisted of sickle cell gene “S” and Haemoglobin “C” gene, while Mrs “D” was Sickle Cell gene “S” and “beta- Thalassaemia” for Sickle cell beta-Thalassaemia disease [19 20]
Figure 3: ACHEACHE x ACHEACHE produce 13 ACHEACHE offspring [19 20]
2. A Ghanaian Physician Specialist married a doctor, and they had 4 children with no health problems whatsoever. They thought they had “finished”, but 5 years after the last child what they called “an unplanned pregnancy” occurred and they had a lovely little girl. By the age of 12 months the child began to have signs and symptoms of what they both knew implied sickle cell disease (hand-foot syndrome). They realized they must both have been carrying sickle cell trait NORMACHE (“AS”) and never tested themselves before. Although the girl was lovely, and was indeed the most brilliant of all their children they said they found the need for Genetic Counselling and Voluntary Family Size Limitation even when couples did not suspect they carried Sickle Cell Trait.
3. This “T-S” Kwahu family knew they were both sickle cell trait NORMACHE “AS” and said they were told that 1 in 4 of any children they had would be “SS” ACHEACHE. They thought that was fair enough Genetic Gambling, and as they were tremendously in love they went ahead to produce a family. When first child, a boy, was “SS” they thought, they could have more children because the 1 in 4 had already come, and they were looking forward to the following 3 children without the ACHEACHE combination. Read the story in my book “The Sickle Cell Disease Patient”, pages 500 & 501: Second child, boy, “SS”, third “SS”, fourth “SS”, fifth “SS” and sixth “SS” [2]. But why did they continue having so many children? Answer: Because the only girl among the 6 children had died young, and Mrs T-S desperately wanted a girl. Moral of this true story is that it is wrong to tell people that Genetic Gambling will produce the different phenotypes in a certain order. As the first 4 children of the NORMACHE doctors mentioned above turned out without being ACHEACHE phenotypes so it does happen that other NORMACHE parents have been known to produce children all of whom are ACHEACHE – the term gambling will not have any relevance otherwise. The “1 in 4” prediction advice is deceptive and must be pointed out in any competent genetic counseling exercise.
4. Male Procreation needs to be curbed as part of Genetic Counselling and Voluntary Family Size Limitation (GCVFSL). On Saturday June 19 1971, Mr Kofi Sampani pulbished in The MIRROR (Accra) a front page story from Sunyani of Okomfo Klutse with a picture of his 23 wives who produced 94 children [21]. Okomfo Klutse could be NORMACHE, or NORMNORM or ACHEACHE “SC”, and no less than 8 of his wives would be carrying an ACHE gene. If he was ACHEACHE then all 94 children would have one of his ACHE genes, and any of the wives who gave an ACHE gene would have a child or children suffering from Ahotutuo. If this man was NORMACHE, like my own father was, then 47 of his children would receive an ACHE gene, and some of the children would be ACHEACHE that resulted from some of the 8 wives passing on their ACHE genes. A year earlier in 1970 I had said in the Journal of Tropical Medicine and Hygiene in England that polygamy could increase the burden of sickle cell disease [22], so males with the Trait (NORMACHE) should limit family size because of their high MPSI (Male Procreative Superiority Index) a genetic index that I invented [2 15-17] to prove that geneticists must not say that Malaria/Sickle Cell Trait Balanced Polymorphism was the main reason for the high incidence of the sickle cell trait in African populations. [23 24]. Professor George Bonney and I showed mathematically the role of polygamy in Population Genetics [25]. Incidentally, there is a high incidence of sickle cell trait among white populations in Greece, Turkey, and India [13 26].
OUR DUTY FOR THE PRESENT AND FUTURE
Populations all over the world are looking for governments to solve their health problems for them. We must be careful not to extend this “Government will do it all for me” attitude to Genetic Disease. Governments can make it easier for each of us to find out whether we are NORMACHE or NORMNORM or ACHEACHE to determine what exactly does our ACHE represents: “S”?, or “C”?, or “F-hereditary”?, or “beta-Thalassaemia”? or “Haemoglobin Korle Bu”? or Haemoglobin Osu-Christiansborg?”, or “K”?, or “D”?. True, our governments can help us [27], but we need ourselves to do something about our health, and our children’s health. My Distinguished Professorship advises you thus: “DO IT YOURSELF!”
Figure 4: Service, Education, and Research Dimensions of Genetic Counselling
This SCHEMA is the way forward. Let us know that if we are NORMNORM our grand children could be ACHEACHE. How is this possible? ANSWER: Because our husband, or wife may be a trait carrier – quite harmless – your son or daughter with the Trait may marry another Trait, like my parents did, and your grand child may then become ACHEACHE. This reasoning is quite simple, so please take this Genetics on board. As I have implied earlier it is no business of Genetic Counselling to ask a NORMACHE never to marry another NORMACHE, or to tell people how many children to have, or to tell them that the chances of hereditary disease in the offspring are such and such. No, my business as a Counsellor is to tell people the facts about the illness, and in this I seek the help of those who have ACHEACHE and who have become great Achievers in various fields of endeavour despite their medical history, and to ask them to join me in advising people to find out what phenotype they belong to, and then put to them their options: (1) Go in for Genetic Gambling, that is the “Probability” route, (2) Go in for the “I have made up my mind to avoid ACHEACHE” route which can be done by using the kanad – the Konotey-Ahulu Norm Ache Dice – to predict with 100 per cent certainty which pairings will always, without fail, avoid the ACHEACHE burden. This involves some painful decisions but has to be done, especially in the UK where many ACHEACHE patients have died from being given Morphine and Diamorphine (Heroin) for sickle cell pain [28 29 30]. Ghanaians have protested about this [31-37], but the Opiods (Opiates) for pain in sickle cell crisis have been officially sanctioned for the UK [38].If you are going to bring up children in the UK, and there is the possibility that if you have more children they will take their parents’ ACHE genes then take note that when they develop pains they will be put on Heroin and Morphine. Think seriously of limiting your family size even when you are most anxious to have another child.
A warning here for Ghanaian Doctors: Please do not adopt British or American methods of managing sickle cell disease when Ghana has produced the largest number of ACHEACHE Achievers in the world [39] without using Morphine or Diamorphine or Hydroxyurea or monthlty blood transfusions as they do abroad.
EXTEND UCC’s ORIGINALITY
I heartily congratulate The University of Cape Coast at their 50th Anniversary for evidences of their Originality – thinking what has not been thought before, saying what has not been said before, and doing what has not been done before. Ayenyekoo! – The plural for “Well Done!” Please extend your Originality to advising every student, teacher, administrative staff, and their families to find out (at their own cost) what their haemoglobin phenotype is. Let this University be the First in the world to have this policy. “At their own expense” is always better because it separates the really serious from the rest. “Save some money to enable you test yourself and your present family, and potential family” must be a Motto. One of the reasons for my KÁGÈ SICKLECELL FOUNDATION (KÁGÈ is Konotey-Ahulu Genetic Epidemiology) [38] is that governments every where have a great reluctance in dealing with genetic disease (except to decree that pregnancies should be terminated), and I want a Foundation that lays emphasis on teaching and on ethical matters in prevention processes. I have been spreading the message in lectures and international medical journals, and I have a website that is consulted around the clock from around the world [14] Since the Year 2000 when the University of Cape Coast made me The Kwegyir Aggrey Distinguished Professor of Human Genetics I have by August 2014 published 172 articles [40] most of which can be Googled and read on line, and all of which include the University of Cape Coast address, emphasizing ethical practice in Human Genetics and Clinical Medicine. May this University go from strength to strength, and may we thank GOD and take courage.
ACKNOWLEDGEMENTS
University of Cape Coast: I thank Vice Chancellor Rev Professor Samuel Adjepong and University Council, and subsequent Vice Chancellors Rev Professor Emmanuel Adow Obeng, Professor Jane Naana Opoku-Agyemang, Professor D D Kuupole, Pro-Vice Chancellors Professor Haruna Yakubu and Colleagues, and Registrars Mr Kofi Ohene and Colleagues of the Vice Chancellor’s Office and the remarkably cooperative Deans of the Faculty of Science since Year 2001 Professor Kobina Yankson, Professor Victor Gadzekpo, Professor Samuel Yeboah-Mensah, Professor B K Gordor, Professor Basuah, and their extremely hardworking Faculty Officers Rev Evelyn Nickel, Mr P K Mensah, Dr Alexis Akanson, Mr Barsi-Serbeng, Mr Ebenezer Aggrey, Ms Mary Obimpeh and their Administrative Staff Ms Emma Bedzo, Mrs Marian Aidoo, Ms Ernsetina Pokua all of whom supervised the Annual Dr Kwegyir Aggrey Prize Exams most admirably. Encouragers were Professors Jeurry Blankson, Eric Anum, Lawrence Owusu-Ansah, John Micah, and Mr Daniel Essel, Mr John K Edumadze, and Mr Sitsofe Tettey.
Indebted I am to a multitude – I am most grateful to the following who were generous in their help, financial and otherwise: Adomako, Albert & Fitnat; Acquaah-Harrison, Asabea; Chapman Nyaho, Mawunu; Akufo-Addo, Edward & Mrs Irene; Owusu, Maxwell; Botchway, Dr Kwesi; Vanderpuije Nii Oti Ahmed and Mrs Risikatu; Firth Drs Peter and Jean; Chew Dr George and May; Tettey Rev Ben and Mrs Margaret; Draisey Dr Mike; Norris Dr Geoff; Djabanor Dr Frank and Mrs Amelia; Matekole Professor Mike Ossom; Archampong Professor E Q; Addy Professor Jonathan H; Asirifi Professor Yaw; Laryea Mr Reginald; Amanor-Boadu, Dorothy; Thompson, Mrs Rosemary; Bruce-Tagoe, Professor Alexander; Codjoe, Lawyer Raymond; Mould-Iddrisu, Mrs Betty; Ekem, Dr Ivy; Tsikata, Capt Retired Kojo; Allotey, Professor Francis; Djangmah, Professor Jerome; Wosornu, Professor Lade; Hamidu, General (Retired) Joshua; Senanu, Professor Kojo; Perbi, Reindorf B; Konotey-Ahulu, Dawid & Rachel; Konotey-Ahulu, Mrs Rosemary; Madubunyi, Boniface; Awoonor, Professor Kofi; Baffour Ankomah; Nene Sakite II, Konor of Manya Krobo; Odoteye Samuel; Gbedemah, Mensah and Sophie; Omaboe, Nortey; Omaboe, Mrs Letitia; Ampim, Kofi & Deborah; Djamson, Dr Eric and Mrs Gladys; Shoetan, Samuel & Cecilia; Bonney, Professor George and Mrs Efua; Dodoo, Professor Benjamin; Awwad, Mr Awwad FRCS; Tetteh, Richard; Savage, Hugh; Badger-Laryea, Ms Christiana; Gorbett, Miss Ann; Bukar, Alhaji Ibrahim; Falae, Mr Samuel; Madueke, Mr Chike; Frimpong-Boateng, Professor Kwabena; Quartey, Dr Samuel (Philadelphia); Angelou, Professor Maya; Kofi, Tetteh J; Mlapa, Joel; Whest, Mrs Adelaide; Kofi-Tsekpo, Professor Mawule; Thairu, Professors Kihumbu and Wanja; Mensah Dr Justice Thomas and Mrs Akosua; Alexandra Road Congregational Church Hemel Hempstead, Herts, England; Ghana College of Physicians and Surgeons (Rebecca Amoh, Professors Paul Nyame, David Ofori-Adjei, J T Anim, Alex Bruce-Tagoe); Ghana Academy of Arts & Sciences (Presidents F T Sai, Letitia Obeng, R F Amonoo; F K Allotey); Academy for the Developing World (TWAS – Triest, Italy, Professor Mohamed Hassan); African Academy of Sciences (Nairobi – Professor Eng. Shem Arungu-Olende).
REFERENCES:
1 Konotey-Ahulu, FID. Aggrey of Africa: “Only the best is good enough for Africa”. New African, October 2004, pages 50-51
2 Konotey-Ahulu, FID. The Sickle Cell Disease Patient. Macmillan Education Ltd London 1991/1992. Foreword by Roland B Scott, MD (HowardUniversity) – 36 chapters with 4,500 references, 643 pages. ISBN: 0-333-39239-6 [Also Tetteh-A’Domeno Company (T-A’D Co 1996) UK – P O Box 165 Watford WD17 3ZH]
3 Konotey-Ahulu FID. The Human Genome Diversity Project: Cogitations of An African Native. Politics and the Life Sciences (PLS) 1999, Vol 18: No 2, pp 317-322. [Invited Commentary on Professor David Resnik’s article: The Human Genome Diversity Project: Ethical Problems and Solutions.] PMID: 12561789
4 Konotey-Ahulu, FID. Sickle Cell Disease. The Case for Family Planning. Accra, Astab Books Ltd, 1973; 32 pages.
5 Konotey-Ahulu, FID. Survey of Sickle Cell Disease in England and Wales. British Medical Journal 1982; 284(6309): 112 doi: 10. 1136/bmj.284/6309/112a Jan 9 1982
6 Konotey-Ahulu FID. The spectrum of phenotypic expression of clinical haemoglobinopathy in West Africa. New Istanbul Contribution to Clinical Science 1978 Dec; 12(3-4): 246-257. PMID: 756535 [PubMed – indexed for MEDLINE]
7 Ringelhann B, Konotey-Ahulu FID, Lehmann H and Lorkin PA. A Ghanaian adult, Homozygous Persistence of Foetal Haemoglobin and Heterozygous for Elliptocytosis. Acta Haematologica (Basel) 1970; 43(2): 100-110. PMID: 4986189 [PubMed – indexed for MEDLINE] http://lib.bioinfo.pl/pmid:4986189
8 Ringelhann B, Acquaye CTA, Oldham JH, Konotey-Ahulu FID, Yawson G, Sukumaran PK, SchroederWA and Huisman THJ. Homozygotes for the hereditary persistence of fetal haemoglobin: The ratio of G to A chains and biosynthetic studies. Biochem Genet 1977 Dec; 15(11-12): 1083-1096. PMID: 603615 [PubMed – indexed for MEDLINE
9 Acquaye CTA, Oldham JH and Konotey-Ahulu FID. Blood-donor homozygous for hereditary persistence of fetal haemoglobin. Lancet 1977 April 9; 1(8015): 796-797. PMID: 66588 [PubMed – indexed for MEDLINE]
10 Ringelhann B, Konotey-Ahulu RID, TalapatraNC, Nkrumah FK, Price B and Lehmann H. Haemoglobin K Woolwich ( 2 2 132lys –> Gln) and its combination with Hb.S and C in two Ghanaian tribes. Acta Hematologica 1971; 45(4): 250-258. PMID: 4999133 [PubMed – indexed for MEDLINE] http://lib.bioinfo.pl/pmid:4999133
12 Konotey-Ahulu FID, Kinderlerer, JL Lehmann H and Ringelhann B. Haemoglobin Osu-Christiansborg. A new chain variant of Haemoglobin A (beta 52 D3 Aspartic Acid –> Asparagine) in combination with Haemoglobin S. Journal of Med Genet 1971 Sep; 8(3): 302-305. [PubMed – indexed for MEDLINE] PMCD: PMC 1469179 & PMID: 5097135 http://pubmedcentral.nih.gov/picrender.fcgi?artid=146917&blobtype=pdf
13 Ringelhann B, Konotey-Ahulu FID. Hemoglobinopathies and thalassemias in Mediterranean areas and West Africa: Historical and other perspectives 1910 to 1997 – A Century Review. Atti dell’Accademia dell Science di Ferrara ( Milan) 1998;74: 267-307
15 Konotey-Ahulu FID. Male procreative superiority in African populations: The fact established and quantified and Papp Z (Eds). Medical Genetics, Exerpta Medica 1977; 600-607.
16 Konotey-Ahulu FID. Male procreative superiority index (MPSI): The missing co-efficient in African anthropogenetics. BMJ Dec 20-27 1980; 281(6256): 1700-1702 doi:10.1136/bmj.281.6256.1700 http://www.bmj.com/cgi/reprint/281/6256/1700.pdf
17 Konotey-Ahulu FID. The Male Procreative Superiority Index (MPSI): its relevance to genetical counseling. In FIFTY YEARS OF HUMAN GENETICS A Festschrift and liber amicorum to celebrate the life and work of GEORGE ROBERT FRASER Edited by Oliver Mayo and Carolyn Leach. Wakefield Press 2007 (www.wakefieldpress.com.au)
1 The Parade West, Kent Town, South Australia 5067
18 Konotey-Ahulu FID. The Third International Conference On The Achievements Of Sickle Cell Disease (ACHEACHE) Patients -19th July 2010, International Conference Centre, Accra. Taking part were adult Sickle Cell Disease Patients in various Professions. Chairman was Professor Kofi Awoonor, Chairman of the Council of State, Ghana. The first ever such Conference was in London at the Royal Society of Medicine, 1 Wimpole Street in 1993. The Second was in Accra in 1995.
19 Konotey-Ahulu FID and Ringelhann B. Sickle-cell anaemia, sickle-cell thalassaemia, sickle-cell haemoglobin C disease and asymptomatic haemoglobin C thalassaemia in one Ghanaian family. BMJ 1969 Mar 8; 1(5644): 607-612. doi:10.1136/bmj-1.5644/607
PMID: 5766126 [PubMed – indexed for MEDLINE] March 8 1969
20 Bentsi-Enchill KK, Konotey-Ahulu FID. Thirteen children from twelve pregnancies in sickle-cell thalassaemia. http://www.bmj.com/cgi/reprint/3/5673/762.pdf BMJ Sept 27 1969; 3(5673): 762 doi:10.1136/bmj.3.5673.762 [In Medical Memoranda Sept 27 1969]
21 Sapanin Kofi. Man with 23 wives and 94 children. Okomfo Klutse. The Mirror, Accra, Saturday 19 June, 1971, No. 932, Front Page.
22 Konotey-Ahulu FID. Maintenance of high sickling rate in Africa: Role of polygamy. J Trop Med Hyg 1970 Jan; 73(1): 19-21 (38 references). PMID: 4906442
23 Allison AC. Protection afforded by sickle cell trait against subtertian malarial infection. British Medical Journal 1: 290-294.
24 Konotey-Ahulu FID. Balanced Polymorphism and Hereditary Qualitative and Quantitative Erythrocyte defects. Ghana Med J 1972; 11: 274-285. [Chapter 8 in Ref. 2]
26 Choremis C et al. Blood group of a Greek Community with high sickling frequency. Lancet 1957; 2: 1333-34
27 Boyo AE, Cabannes R, Conley CL, Lehmann H, Luzzatto L, Milner PF, Ringelhann B, Weatherall DJ, Barrai I, Konotey-Ahulu FID and Motulsky AG. Geneva WHO Scientific Group on Treatment of Haemoglobinopathies and Allied Disorders. (Technical Report) 1972; 509:83 pages.
28 Mason S. Enquiry shows poor care for patients with sickle cell disease. British Medical Journal 2008; 336: 1152
29 NCEPOD (National Confidential Enquiry into Patient Outcome and Death). Sickle: A Sickle Crisis? (2008) [Sebastian Lucas (Clinical Coordinator), David Mason (Clinical Coordinator), M Mason (Chief Executive), D Weyman (Researcher), Tom Treasurer (Chairman) info@incepod.org
30 Konotey-Ahulu FID. Poor care for sickle cell disease patients: This wake up call is overdue BMJ Rapid Response May 28 2008 BMJ 2008; 336: 1152 to Susan Mayor “Enquiry shows poor care for patients with sickle cell disease” on National Confidential Enquiry into Patient Outcome and Death (NCEPOD) REPORT “SICKLE: A Sickle Crisis? (2008)
33 Shoetan Cecilia. I lost my Sickle Cell Disease adult daughter minutes after being given Diamorphine intravenously when she could not breathe. British Medical Journal Rapid Response June 3 2008 http://www.bmj.com//cgi/eletters/336/7654/1152-a#196520
38 Konotey-Ahulu FID. Management of acute painful sickle cell episode in hospital: NICE guidance is frightening! British Medical Journal Rapid Response 7 June 2012 http://www.bmj.com//cgi/eletters/344/bmj.e4063?ti
39 Konotey-Ahulu FID. KAGE SICKLE CELL FOUNDATION [Konotey-Ahulu Genetic Epidemiology Sickle Cell Foundation] Launched 2 September 2011 Ghana College of Surgeons, Accra.[Professor Kofi Awoonor as Chairman, and Nene Sakite II, Konor of Manya Krobo as Guest of Honour]
Kwegyir Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL, England
Kwegyir Aggrey Distinguished Professor of Human Genetics University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL, England
———————————————————————————————————-
ABSTRACT
Sickle Cell and Allied Haemoglobinopathy:
The Genetics that touches You and Me
Felix I D Konotey-Ahulu
Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast Ghana and Consultant Physician Genetic Counsellor 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL, England
Originality, without doubt, is a hallmark of the University of Cape Coast. I define Originality as thinking what has not been thought before, saying what has not been said before, and doing what has not been done before. Right from the beginning when Head of State Dr Kwame Nkrumah decided to create a Third University in Ghana national and international eyebrows were raised. But this 50 year’s Anniversary of what skeptics called “an experiment” has proved many wrong because UCC has many things to show that reflect sheer originality. Not least of which was the creation of the First Personal Chair on the African Continent to be linked to the name of an Educational Legend Dr James E Kwegyir Aggrey who thought things that had not been thought before, said things that had not been said before, and did things that had not been done before.
When Vice- Chancellor Rev Professor Samuel Adjepong, with the University Council made me The Dr Kwegyir Aggrey Distinguished Professor of Human Genetics in the University of Cape Coast and at the same time gave me an Honorary Doctorate in Science (when Dr Daniel Chapman Nyaho was also honoured with a Doctorate) I was greatly humbled and extremely grateful. I knew that work displaying originality was expected of me. This chapter contribution to the Golden Jubilee gives a little glimpse of what I have used the University of Cape Coast title to achieve. Quite apart from identifying UCC Science Graduates with genius through the Annual Dr Kwegyir Aggrey Prize Examinations, I have been ceaselessly engaged in skirmishes at the frontiers of Medicine especially where Ethics is made to take a back seat and commercial concerns promoted to the fore at the expense of patients’ health. A 32 year old son of a UCC Professor (Haemoglobin ‘SC’ phenotype) walked into a British Teaching Hospital on a Friday, and by the following Tuesday he was dead on a Heroin drip. A 32 year old Ghanaian lady (also Hb “SC” phenotype) walked into a London hospital, and within 18 hours she too was dead, with her mother, a nurse, looking on while Heroin was injected. I give the British Medical Journals ‘Full Marks’ for publishing not only my informed protests from the UCC platform, but also those of other Ghanaians including the Professor whose son died. You need to pay attention to what I have said here because Haemoglobinopathy affects all of us. Look up all the 40 references.
This article has also been published here on the Cape Coast website http://bit.ly/1DzHceM
Introduction
At least two groups of scientific thinkers or thinking scientists will frown on this article. First, those who base their atheism on reason, and secondly anti-Semitics. My plea is that both these groups – brilliant atheists and brilliant Jew haters read carefully what I have presented here and comment. There are some historic events whose explanation
defies scientific scrutiny [1]. Yet some aspects of these events can, within limits, have scientific explanations. Nobel laureate Sir Peter Medawar is one of the few original thinkers who recognize that there are limits to Science [2]. One such is origins (Ultimate questions) that are “beyond the explanatory competence of science” [2]. Medawar explains what he means by ultimate questions: “That there is indeed a limit upon science is made very likely by the existence of questions that science cannot answer and that no conceivable advance of science would empower it to answer.” [2, page 66]. Scientists can only hypothesize about origins but their theories do not change the fact that origins are historical events. The paradigm which says historical events that cannot be explained scientifically must be deemed not to have happened is contrary to Professor Medawar’s cogitations. And who am I to disagree with him?
History Versus Limits of Science: Is Solomonic Genius a Y Chromosome Phenomenon?
To read more click to open the PDF document above.
Valentine Brousse and colleagues’ Clinical Review [1], gives doctors little guidance for patient management. “Serjeant’s and my combined experience of 80 years covering thousands of patients in sickle cell crisis” can help. [2].
DISAGREEMENT WITH NICE ON PATIENT MANAGEMENT
Graham Serjeant’s Jamaican experience [3] and my Ghanaian [4] differ from NICE’s Guidance [5] in UK where patients died from Morphine/Diamorphine overdose. [6 7] We managed sickle cell disease (scd) patients to university without Morphine/Diamorphine, Hydroxyuurea, or regular transfusions. Our experience (References 1965 to 2014} could save UK patients.
(1) Two brothers and one sister suffered from scd “Hereditary Rheumatism” traced back to 1670 AD www.konotey-ahulu.com/images/generation.jpg [9-11]. Siblings’ commonest cause of sickle cell crises characterized by convulsions and priapism [9 10] was Malaria. One brother had ‘gnathopathy’, my invented word for maxillary marrow hyperactivity [10 12 13 14].
(2) Directing largest Sickle Cell Clinic I catalogued 133 Illustrative Case Histories [10] including alcohol-induced myocardial infarction [10, p 503] and mental nerve neuropathy [15], kanumblll sign. Investigate any numb lower lip for scd. [15]
(3) Haematologist Helen Ranney published “There is no single clinical experience in the United States comparable to that of Dr Konotey-Ahulu” [16].
(4) Receiving with Linus Pauling and others “Martin Luther King Jr Foundation Award for Outstanding Research in Sickle Cell Anaemia” my Keynote Address “Difference between Sickle Cell Disease and Sickle Cell Trait” that exposed fraud of Insurance Companies led to being given 4 bodyguards in Philadelphia. [17] By saying Sickle Cell Trait was “largely asymptomatic” [1] Brousse et al repeated Insurance Companies’ misinformation/disinformation [18-21] used to levy Traits 150% Premium.
(5) On WHO Expert Genetics Advisory Panel with Haemoglobinopathy Greats who valued Ethics in Human Genetics. [22]
LESSONS FROM GRAHAM SERJEANT’S JAMAICAN EXPERIENCE
“In Jamaican experience morphia or its derivatives are rarely used or necessary” [23] and “the most painful crises may be treated in a day centre, the patient returning home in the evening” [24]. See more of Serjeant’s publications from 1968 [23-38].
TIPS FROM MY FIRST PUBLICATIONS IN 1965 [39-41] TO LATEST 2014 [42 43]
(1) KEEP A DIARY! Circumstances [10 39], some patient-specific, precipitate crises eg Ghanaian scd man had sickle crisis on eating oranges [10]. Would Grousse et al start Hydroxyurea [1] after twice eating oranges?
(2) ALWAYS CARRY UMBRELLA. One rain-soaked scd boy got sickle crisis and stroke [44]. Trans-cranial Doppler [1] better prophylactic than raincoat? Hot weather too causes sickle crisis [39].
(3) NOSE PICKING causes Epistaxis [39 45].
(4) VALSALVA MANOEUVRE: In 18 causes of ocular bleeds [10] “six were related to sneezing, blowing nose, shouting, bending down, lifting heavy objects, and vigorous exercise” [46]. Avoid Valsalva in labour; Caesarian Section advised.
(5) DOCTORS BEWARE! Ectopic pregnancy, gall stones [47], appendicitis, splenic infarct [10] misdiagnosed as “Abdominal crisis”. Squatting (Tourniquet effect) can start crisis [10]. Opiates cause “Chest syndrome” [48]. Pulmonary embolism missed [49]. Note Intra-family scd phenotype differences [50 51]
(6) EXAMINE COMMUNITY RUMOUR. Medicated bed nets [1] no better than usual nets and may harm babies [52]. Malaria kills scd patients quicker [10 53-55]. Patients shun Hydroxyurea [56 57] and prenatal diagnosis [58 59] Drinking up to 4 L of water a day has stopped crises for past 25 years, as has “Aerobic Oxygen” 20 drops tds in water [60] Coconut juice clearing jaundice? [60.]
(7) STATIONERY BICYCLE aborts early morning priapism [10, 43]
(8) PRE-SURGERY PARTIAL EXCHANGE TRANSFUSION (Bedside method) [10].
(9) GENETIC COUNSELLING & VOLUNTARY FAMILY SIZE LIMITATION [61-69]
(10) SICKLE CELL DISEASE PATIENT ACHIEVERS teach doctors 1993-2010 [70]
(11) G6PD Deficiency: 1 in 4 male and 1 in 16 female scd patients have this. [71 72]
(12) WORLD SICKLE CELL DAY BROADCASTS AND RESOURCES [73 74]
WHAT CHOICE FOR UK CLINICIANS?
NCEPOD discovered “9 out of 19 patients with sickle cell disease who had pain on admission and who then died had been given excessive doses of opiods” [6 7]. Yet NICE still recommends “a strong opiod intravenously” [5]. But Why? [2 4 10 48 75-87]. Patients complain to BMJ [88-90].
1 Brousse V, Makali J, Rees DC. Management of sickle cell disease in the community. BMJ 2014; 348:g1765 doi:10.1136/bmj.g1765
2 Konotey-Ahulu FID Opiates for sickle cell crisis? Lancet 1998: 351: 1438 “The question that puzzles me is why do west African and West Indian patients with sickle cell disease who did without morphine in our countries have to be given morphine pumps during sickle cell crisis when they come to the UK?”
3 Serjeant G. The case for dedicated sickle cell centres. BMJ 2007; 334: 477
5 NICE GUIDELINES. Management of acute painful sickle cell episode in hospital; summary of NICE Guidance. BMJ 2012; 344: doi:http://dx.doiorg/10 1136/bmj’e’4063
6 NCEPOD – National Confidential Enquiry into Patient Outcome and Death. SICKLE: A Sickle Crisis? 2008 [Sebastian Lucas (Clinical Coordinator), David Mason (Clinical Coordinator), M Mason (Chief Executive), D Weyman (Researcher)/ Tom Treasurer (Chairman) info@incepod.org
7 Mason S. Enquiry shows poor care for patients with sickle cell disease. BMJ 2008; 336: 1152 “In 2 years 9 out of the 19 patients with sickle cell disease who had pain on admission and who then died had been given excessive doses of opiods”
8 Konotey-Ahulu FID. http://bit.ly/1gDdMlN International request to manage patients, for example, sicklecell.md/blog/index.php/2007/06/request-from-geneva-for-patient-in-hospital-in-colorado-usa June 2007
9 Konotey-Ahulu FID. Pattern of Sickle Cell Disease in Ghana (A Study Of 1,550 Consecutive Patients) – A Thesis Presented For The Degree of Doctor of Medicine (M.D.) In The University Of London 1971 Awarded Feb 1972.
10 Konotey-Ahulu FID. The Sickle Cell Disease Patient. Clinico-epidemiological study of 1550 consecutive patients at Korle Bu Hospital, Accra. T-A’D Co, Watford 1996 http://www.sicklecell.mde/aboutscd.asp
(Reprint of Konotey-Ahulu FID. The Sickle Cell Disease Patient. Natural History from a clinico-epidemiological study of the first 1550 patients of Korle Bu Hospital Sickle Cell Clinic. London & Basingstoke, Macmillan Press Ltd 1991/1992. Foreword by Roland B Scott MD, Howard Univ, Washington DC)
11 Konotey-Ahulu FID. Sickle Cell Disease In Successive Ghanaian Generations For Three Centuries (Manya Krobo Tribe) In The Human Genome Diversity Project: Cogitations of An African Native. Politics and The Life Sciences (PLS) 1999; Vol 18: No 2, pp 317-322.
12 Konotey-Ahulu FID. Effect of environment on sickle cell disease in West Africa; epidemiologic and clinical considerations. Chapter 3 in SICKLE CELL DISEASE – diagnosis, management, education and research. Eds Ahramson, Bertles JF, Wethers Doris L; St Louis – CV Mosby Co 1973, pp 20-38.
14 Konotey-Ahulu FID. The liver in sickle cell disease. Clinical aspects. Ghana Med J 1969; 8: 104-118. (First ever use of word “Gnathopathy”)
15 Konotey-Ahulu FID. Mental nerve neuropathy: a complication of sickle cell crisis. Lancet 1972; 2: 388 [Constitutes discovery of a new physical sign in Clinical Medicine – The kanumblll sign spells out who discovered it, what it is, and where published: “konotey-ahulu numb lower lip Lancet” sign 1972]
16 Ranney Helen. Summary of Symposium 1972 on Sickle Cell Disease – Diagnosis, Management, Education and Research – In SICKLE CELL DISEASE, Eds H Abramson, John F Bertles, Doris Wethers (C Mosby Co), 1973, page 320: “There is no single clinical experience in the United States comparable to that of Dr Konotey-Ahulu”
17 Konotey-Ahulu FID. Four bodyguards and the perils of unmasking scientific truths www.bmj.com/cgi/reprint/335/7612/210.pdf BMJ 2007; 335: 210-11 BMJ July28 2007.
18 Konotey-Ahulu FID. Beware of symptomatic sickle cell traits. Lancet 1992; 339: 555. Doi:10.1016/0140-6736(92)90377-F (Pointing out that the Sickle Cell Trait ‘AS’ may not be true Sickle Trait, and that the Haemoglobin C Trait ‘AC’, may be something else, as the “A” has been shown could stand for Hb Quebec-Chori, by Witkowska et al – New Eng J Med 1991; 325: 1150-54 http://www.thelancet.com/journals/lancet/article/PII0140-6736(92)90377-F/fulltext
20 Konotey-Ahulu FID. Sickle cell Trait Misinformation and Disinformation Nov 30 2011 www.sicklecell.md/blog/?=108 [Comprehensive Review]
21 Konotey-Ahulu FID. Further Communication on “Sickle Cell Trait Misinformation and Disinformation” and Sickle Cell Terminology: Disease or Disorder? www.sicklecell.md/blog/?p=127 April 16 2012
22 Boyo Alex E, Cabannes R, Conley CR, Lehmann H, Luzzatto L, Milner PF, Ringelhann B, Weatherall DJ, Barrai I, Konotey-Ahulu FID, Motulsky AG. WHO (Geneva) Scientific Group on Treatment of Haemoglobinopathies and Allied Disorders. (Technical Report) 1972; 509: 83 pages.
23 Serjeant GR. Sickle Cell Disease. Oxford, Oxford University Press, 1985.
24 Serjeant GR. Sickle cell disease. Lancet 1997; 35: 725-730.
25 Serjeant GR, Richards R., Barbor PRH, Milner PF. Relatively benign sickle cell anaemia in 60 patients aged over 30 in the West Indies. BMJ 1968; 3: 86
26 Serjeant GR, Serjeant BE, Milner PF. The irreversibly sickle cell: a determinant for haemolysis in sickle cell anaemia. Br J Haematol 1969; 17: 527-533.
27 Serjeant GR, Galloway RE, Gueri MC. Oral zinc sulphate in sickle cell ulcers. Lancet 1970; 2: 891
28 Serjeant GR. The clinical picture of sickle cell anaemia in Jamaica. MD Thesis, University of Cambridge, 1971.
29 Serjeant GR, Ashcroft. Shortening of the digits in sickle cell anaemia. A sequel of the hand-foot syndrome. Trop Gegraph Med 1971; 23: 341-346.
30 Serjeant GR, et al 1972. The conjunctival sign in sickle cell anaemia. JAMA 1972; 219: 1428-31
31 Serjeant GR, et al. The clinical features of sickle cell beta-thalassaemia in Jamaica. Brit J Haematolol 1973; 24: 19-30
32 Serjeant GR, et al. The clinical features of haemoglobin SC disease in Jamaica. Brit J Haematolol 1973; 24: 491-500.
33 Serjeant GR et al. Screening of cord blood for the detection of sickle cell disease in Jamaica. Clim Chem 1974; 20: 666-69.
34 Serjeant GR et al. The internal auditory canal and sensori-neural hearing loss in homozygous sickle cell disease. J Laryngol Otol 1975: 89: 453-455
38 Serjeant GR. Blood transfusion in sickle cell disease. A cautionary tale. Lancet 2003; 361: 1659-60 [Graham Serjeant’s adult not-transfused patient in Jamaica went on holiday in USA, was transfused and died!].
39 Konotey-Ahulu FID Konotey-Ahulu FID. Sicklaemic human hygrometers. Lancet 1965; 1:1003 1004 [Listing precipitating causes of crises eg hot weather etc] [http://www.pubmedcentral.nih.gov/picender.fcgi?artid=1846286&blobtype=pdf
40 Konotey-Ahulu FID. Torrential epistaxis associated with symmetrical facial skin ulceration in sickle cell anaemia. BMJ 1965; 2:859-860 doi:10.1136/bmj2.5466.859 http://www.bmj.com/cgi/reprint/2/5466/859.pdf
46 Konotey-Ahulu FID. Valsalva vitreous haemorrhage and retinopathy in sickle cell haemoglobin C disease. Lancet 1997; 349: 1774
47 Archampong EQ, Konotey-Ahulu FID. Biliary tract disease and sickle cell anaemia in Korle Bu Hospital, Accra. Ghana Med J 1975; 14: 176-180
48 Ringelhann Bela, Konotey-Ahulu FID. Haemoglobinopathies and Thalassaemias in Mediterranean areas and in West Africa: Historical and other perspectives 1910 to 1997. Accademia del Scienze Ferrara Atti, volume 74, Anno Accademico 174 1996-97, pages 297-307 [A Century Review]
50 Konotey-Ahulu FID, Ringelhann B. Sickle cell anaemia, sickle cell thalassaemia, sickle cell haemoglobin C disease, and asymptomatic haemoglobin C thalassaemia in one Ghanaian family. BMJ 1969; 1: 607-612. http://www.bmj.com/cgi/reprint/2/5648/48.pdf [4 Hbnopathy phenotypes]
51 Konotey-Ahulu FID. Patterns of clinical haemoglobinopathy. East African Med J 1969; 46: 149-156 (With tables that distinguish phenotypes clinically)
52 The Globe and Mail, Toronto. Tough flights for mosquito nets. “If they are safe for babies and mothers in Africa, why are they not safe enough in Canada for a week?” 18 August 2006.
54 Djabanor FFT, Reindorf CA, Konotey-Ahulu FID. The effect of sickle cell disease on Ghanaian children. In First International Publication No (HSM) 1974; 73-9141: 70-87
56 Olujohungbe A, Cinkotal I, Yardumian A. Hydroxyurea therapy for sickle cell disease in Britain. BMJ Editorial 1998; 316: 1689. “Many patients are unwilling to take the drug.”
57 Olujoungbe Ade. Bi-directional trust is needed in pain management in sickle cell disease. BMJ Rapid Response 2 July 1999 to Maxwell K, Streetly A, Bevan D BMJ 1999; 318: 1585-1590.
59 Konotey-Ahulu FID. Refusing to provide a pre-natal test for refusing later termination of pregnancy; can it ever be ethical? BMJ Rapid Response. November 20 2006 http://www.bmj.com/cgi/eletters/333/7577/1066#149662
60 Konotey-Ahulu FID. The Sickle Achievers (1). Ghanaian Times, July 23 2005 “I cannot think of a single Ghanaian family that did not have or know of someone with sickle cell disease – known by Tribal names”
61 Konotey-Ahulu FID. The Sickle Achievers (2) Ghanaian Times. August 13 2005 “Some time ago I coined the term for cold season Rheumatism which Europeans call Sickle Cell Disease … The ACHEACHE Syndrome (1 ACHE from each parent”. Makes it easy for Genetic Counselling.
62 Konotey-Ahulu FID. Genetic Counselling in sickle cell disease. BMJ 1969; 3: 235 http://www.bmj.com/cgi/reprint/3/5664/235.pdf (Put ‘ACHE’ for ‘BAD’) PERSONAL VIEW doi:10.1136/bmj.2.5648.48
63 Konotey-Ahulu FID. Sickle cell disease. The Case for Family Planning ASTAB Books Ltd, Accra, 1973
65 Ringelhann B, Konotey-Ahulu FID, Yawson G, Bruce-Tagoe AA, Miller A, Huisman THJ. Alpha Thalassaemia in West Africa. Symposium in Medical Genetics, Debrecen-Hajduszoboslo, Hungary (April 26-29 1976), pp 614-616 in Szabo G and Papp Z, Eds Medical Genetics, Excerpta Medica 1977
66 Konotey-Ahulu FID. Maintenance of high sickling rate in Africa: Role of polygamy. J Trop Med Hyg 1970 Jan; 73(1): 19-21 (38 References)
69 Konotey-Ahulu FID. The Male Procreative Superiority Index (MPSI): It’s relevance to genetical counselling. In FIFTY YEARS OF HUMAN GENETICS – A Festschrift and liber amicorium to celebrate the life and work of GEORGE ROBERT FRASER. Ed: Oliver Mayo & Carolyn Leach. Wakefield Press 2007, 1 The Parade West, Kent Town, Sth Australia www.wakefieldpress.com.au
70 Omaboe Letitia, Konotey-Ahulu FID. The Second International Conference on The Achievements of Sickle Cell Disease Patients. Accra 19th July 1995 Conference Brochure
71 Acquaye CTA, Gbedemah KA, Konotey-Ahulu FID. Glucose-6-phosphate Dehydrogenase Deficiency Incidence in Sickle Cell Disease patients in Accra. Ghana Med J 1977; 16: 4-9
72 Konotey-Ahulu FID. G6PD Deficiency in Ghanaians. How to recognise it.
Click ‘BLOG’ on www.sicklecell.md and click on January 2008 for 20 answers.
73 Konotey-Ahulu FID. WORLD SICKLE CELL DAY 19th June 2012 www.sicklecell.md/blog/?p=132 Featuring (i) The Inheritance of Sickle Cell Disease (ii) The Person with Sickle Cell Disease (iii) The Teenager with Sickle Cell Disease (iv) The Adult with Sickle Cell Disease.
74 WORLD SICKLE CELL DAY 19th June 1013. Broadcast Interview by Tunu Louise Roberts emphasising Public Health Approach to management, and Genetic Counselling with NORMACHE as Trait and ACHEACHE as Disease www.sicklecell.md or www.konotey-ahulu.com (Home Page) Also accessed as http://youtu.be/wEyebVIhr7Q [Suitable for patients and parents
75 Konotey-Ahulu FID. Morphine for painful crises in sickle cell disease. BMJ 1991; 302: 1604 (Commenting on Professor Chamberlain’s recommendation of morphine in pregnancy in sickle cell disease – BMJ 1991; 302: 1327-30). “In obstetrics what happens too foetal respiration when morphine is used?” http://www.bmj.com/cgi/reprint/302/6792/1604-c.pdf
76 Konotey-Ahulu FID. Management of patients with sickle cell disease. African Journal of Health Sciences 1998; 5: 47 [On Sally Davies and Lola Oni in BMJ 315: 656-60 “The Central Middlesex management protocol uses morphine infusions”] Response: “I fear Davies and Oni’s statement will make morphine the accepted drug for sickle crisis management. The consequences for such an approach are dire, especially when some UK hospitals are already making diamorphine their first choice”. And what did NCEPOD find in 2008?
82 Konotey-Ahulu FID. Poor care for sickle cell disease patients: This wake-up call is overdue. BMJ Rapid Response (May 28) BMJ 2008; 336: 1152 http://www.bmj.com/cgi/eletters/336/7654/1152-a#196244 to Susan Mayor’s “Enquiry shows poor care for patients with sickle cell disease” on National Confidential Enquiry into Patient Outcome and Death (NCEPOD) REPORT “SICKLE: A Sickle Crisis? (2008)” http://www.info@ncepod.com
84 Konotey-Ahulu FID. UK Drug related deaths are still rising: So where is NICE? http://www.bmj.com/cgi/eletters/sep01_1/b3536#219836 BMJ Rapid Response to S Mayor: “UK drug related deaths are still rising.” Sept 6 2009. .
86 Konotey-Ahulu FID. Management of an acute painful sickle cell episode in hospital. NICE Guidance is frightening! BMJ Rapid Response Sept 7 2012 www.bmj.com/content/344/bmj.e4063/rr/599158 (42 References)
87 Konotey-Ahulu FID. Opiods for chronic non-cancer pain – Chemotherapy – Clinical Guidelines: Where does ultimate responsibility lie? BMJ Rapid Response www.bmj.com/content/346/bmj.f2937/rr/651421 June 25 2013
88 Dankwa Akosua M. Sickle Cell patients deserve to live. BMJ Rapid Response to NCEPOD Report 11 July 2008 [Adult “SS” wrote to the BMJ: http://www.bmj.com/cgi/eletters/336/7654/1152-a “I know 60 and 70 year olds (yes, sickle cell patients) who have got to that age without ever receiving this as treatment”.
89 Chapman Nyaho Mawunu. Poor care for the sickle cell disease patient: “Pain won’t kill him, but Morphine could”. BMJ Rapid Response to NCEPOD by adult Sickle cell haemoglobin C disease grand-mother 17 June 2008 http://www.bmj.com/cgi/eletters/336/7654/1152a
90 Amanor-Boadu Dorothy, Bruce-Tagoe Alexander, Konotey-Ahulu Felix. The Third International Conference On The Achievements Of Sickle Cell Disease (ACHEACHE) Patients, Accra – 19th July 2010. Adeko Ltd, Accra ISBN: 978-9988-1-3927-8 “One known ‘SS’ man with a PhD, who had never in his 63 years been transfused though Hb level was never above 8.8 g/dL and who had never been prescribed Hydroxyurea, astonished delegates when he announced ‘I do not remember when I last took a pain killer for my sickle cell anaemia. Drink plenty of water, avoid malaria, and have a positive attitude to life’” [page 15] Quoted in full in link of Reference 85 above. [Dorothy Amanor-Boadu herself is a 59-year old “SS” Nurse Oncologist in Accra].
Competing interests: Coming from a family with 3 Sickle Cell Disease (scd) siblings I find it very difficult to remain detached when the received wisdom in the management of scd patients that was proven by NCEPOD to be harmful is still endorsed by NICE.
FELIX ID KONOTEY-AHULU, Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast, Ghana
Consultant Physiician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathies, 9 Harley Street Ltd., Phoenix Hospital Group, London W1G 9AL
President J F Kennedy Assassinated Friday November 22 1963
Westminster Chapel with Dr Martyn Lloyd-Jones Sunday pm Nov 24 1963
Diary Entry of Dr Felix I D Konotey-Ahulu in London
LORD’s Day 24th Nov 1963: The Doctor preached as usual on John 1 v 16 in morning, and Gal. 6 v 14 in evening and he made it most relevant to President Kennedy’s assassination – The Cross as the only thing that makes peace, and brings people together. “I would wish President Kennedy’s murder to bring peace, but I know it won’t. I tell you of a MURDER that reconciles sinful man to GOD!” It wastremendous. Spent day at Mum’s with my beloved & Dawid. Evening prayer together. We heard after chapel the suspect of Kennedy’s killer shot dead.
——————————————————————————————-
On the 50th Anniversary, our Baby Dawid who was then 3 months old when JFK was killed, has now reproduced the entire Sunday evening Service at Westminster Chapel in which the words of the preacher, Dr Martyn Lloyd-Jones, quoted in bold letters above were said. See www.soundcloud.com/dawid1 and click on JFK, and join those of us who were there half a century ago in a Remembrance Worship:-
Prayer (Short)
Hymn: Before Jehovah’s awful Throne
Scripture Reading: Ephesians 2 verses 1 to 22
Hymn: My GOD I thank Thee
Prayer (Comprehensive)
Hymn: In the Cross of Christ I glory
Sermon: Galatians 6 v 14 “God forbid that I should glory save in the Cross of Christ”
Professor George Ebow Bonney, Ph.D. Research Scientist, Musician, and Christian Tribute by Professor Felix I D Konotey-Ahulu*
He was a genius! George Ebow Bonney could have read any subject in university and been able to excel at whatever he did. He chose Mathematics with special emphasis on Statistics, making a huge impact on both sides of The Atlantic. It was my great fortune to have had George work with me at the erstwhile Ghana Institute of Clinical Genetics at the Korle Bu Teaching Hospital in Accra.
RESEARCH
Using students on summer holiday from the then 3 main universities (Legon, Cape Coast, and Kumasi) and paying them handsomely I had organized a countrywide survey of male/female differential procreation to decide how this affected the prevalence of the Sickle Cell Gene in the various regions in Ghana. We had just found that males, indeed, had more children than females when George joined us on the advice of geneticist Professor Ebenezer Laing of the University of Ghana. “George”, I said to him “I want a mathematical underpinning of this fact known to all Africans, but which Europeans appear to think is nonsense because they cannot imagine how a man can have more children than a woman”. I presented him with our data, and in no time George produced a marvelous article which we published in the world’s leading Science magazine with him as senior author entitled: “POLYGAMY AND GENETIC EQUILIBRIUM” We travelled to Debrecen in Hungary to present papers at the International Congress in Human Genetics.
We next embarked on Twin Studies from 13,000 consecutive births at the Korle Bu Teaching Hospital. Our unique findings that in Ghana 1 in every 30 consecutive deliveries produced twins compared with 1 in 80 single births in Britain, 1 for 86 in the USA, and 1 in 145 in Japan enthralled geneticists. George Bonney was the one to present the findings of our team (Mary Walker, Koblah Gbedemah, and me) at the International Congress on Twin Studies in the USA, available today as Proceedings of the Second International Congress on Twin Studies Part C, Editor Walter Nance in Progress in Clinical and Biological Research 1978; Volume 24 Pt B, pages 105 to 108.
George Bonney was simply, simply brilliant. When he crossed the Atlantic his worth was soon recognized. His field of research broadened from Genetic Epidemiology, to Statistical Analysis, Mapping of Human Chromosomes, Breast Cancer, Alcoholism, Nicotine Dependence and, recently, Prostate Cancer. Louisiana State University and Howard University held him in very high esteem. Teamed up with Professor Georgia Dunston and brilliant Others at the Howard University Genome Project, George Bonney’s input was well acknowledged. I myself benefited much when he and Professor Dunston invited me to contribute to the First International Inaugural Conference on The Human Genome in Washington DC in 2009. The measure of his global impact can be gauged from the 1988 Tenth Annual George W Sender Award for (wait for it) “The best published work in Biometry in any journal sponsored jointly by the American Statistical Association & the Biometrics Societies of North America” [For researchers seeking to check on this see (i) “Regressive logistic models for familial disease and other binary traits” Biometrics 1986; Volume 42: pages 611 to 625, and also (ii) Biometrics 1987; Volume 43: pages 951 to 973]. He published right up to 2012.
In one National Award Ceremony in the USA, when his name was announced to receive First Prize the audience was expecting to see a Japanese walk to the podium (the name “Bonney” sounded rather Japanese), and were astonished to behold a Ghanaian African walk up accompanied by great applause. I have quite deliberately gone into some detail of what George Ebow Bonney achieved in Science not only to encourage Africa’s younger generation to emulate his hard work, but also (as we shall see in the next section) to debunk the present all-pervading aggressive atheism that says brilliant people do not accept the existence of GOD.
GEORGE BONNEY’S FAITH EXPRESSED IN MUSIC
George’s musical proclivities became evident when, at the University of Ghana, he directed the Male Voice Choir of the University Christian Union. He played the piano and the organ with virtuosity, and his detailed knowledge of Tonic Sol-fa made it easy, and a joy, for him to teach all 4 vocal parts to singers. Remarks Reindorf Baah Perbi, himself an attractive bass voice singer: “I think it was in the Male Voice Choir that some of us learnt the song ‘When we all get to Heaven’” the first verse with chorus of which goes:
Sing the wondrous love of Jesus
Sing His mercy and His grace
In the mansions bright and blessed
He’ll prepare for us a place.
When we all get to heaven
What a day of rejoicing it will be! When we all see Jesus We’ll sing and shout the victory!
But Joanna Nerquaye-Tetteh quickly adds that it was not just the male choir that George Bonney taught sacred music to: “He taught the Female Voice Choir too, and he taught us to sing ‘It’s not an easy road, but The LORD is with us!’”
Capt. James Hackman Tachie-Menson’s Hymnal and Book of Anthems
To me Professor George Bonney’s musical legacy is entwined with that of Capt. James H Tachie-Menson whose 4 books are a MUST READ and a MUST SING by all, and I mean all, Ghanaians. I am constrained (if I am to do justice to the sweet memory of George Bonney) to quote the first paragraph of his Foreword to the four extraordinary books:“The four volumes of Songs from Land and Sea: The Captain’s Hymnal in Staff Notation, The Captain’s Hymnal in Tonic Sol-fa Notation, The Captain’s Book of Anthems in Staff Notation, and the Captain’s Book of Anthems in Tonic Sol-fa Notation, bring together for the first
“The four volumes of Songs from Land and Sea: The Captain’s Hymnal in Staff Notation, The Captain’s Hymnal in Tonic Sol-fa Notation, The Captain’s Book of Anthems in Staff Notation, and the Captain’s Book of Anthems in Tonic Sol-fa Notation, bring together for the first time, the sacred compositions of Captain James Hackman Tachie-Menson. I have had indescribable joy collaborating with Captain to produce the First Editions of his work.”
For George Bonney to manage to render each (and every one) of these compositions into Tonic Sol- fa, not only for Treble, but also for Alto, Tenor, and Bass is a mark of rare genius. I do not know of any Professor of Music anywhere in the world who could have done that. Captain James H Tachie-Menson says it all in his Preface to the books:
“I would like to give my very special and sincere thanks to Professor George E. Bonney of Howard University in Washington D.C., the originator and coordinator of the project, who of his own volition and initiative personally transcribed the music from staff notation to tonic sol-fa. He has worked tirelessly to bring this project to fruition”.
GIVEN TO HOSPITALITY
George Ebow Bonney was blessed by the Lord Jesus Christ in the gift of Ewura Efua, that wonderful wife of his. What a privilege for Rosemary, my wife, and myself to be very welcome guests in their beautiful home in Rockville, Maryland in the USA. Many others have testified to their hospitality. We saw and profited from their love for The Lord Jesus who equipped Ewura Efua to live with the punishing schedule of her husband who worked long hours, and appeared to do many things simultaneously. Oh how George will be missed!
My heartfelt Condolences and Rosemary’s go to gallant Ewura Efua, and their children and families Aba Bonney Kwawu and Erwin Kwawu and their children Sela Kwawu and Eli Kwawu, and Kofi Bonney and wife Nicole Bonney, and the larger families in Ghana. May the GOD of all comfort surround them with His amazing love in The Lord Jesus Christ through the power of The Holy Spirit. [2 Corinthians chapter 1 verses 3 & 4]. As for George Ebow Bonney, what a Prospect!
H E A V E N
Oh think to step ashore, and that shore Heaven; To take hold of a hand, and that hand God’s hand; To breathe a new air, and that air Celestial air;
To feel invigorated, and know it, Immortality; Oh think, to pass from the storm and tempest To one unbroken smile,
To wake, and find it GLORY!
*Felix I D Konotey-Ahulu is Dr Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathies, 9 Harley Street Ltd, Phoenix Hospital Group, London W1G 9AL [ www.konotey- ahulu.com & www.sicklecell.md ]
Most religious followers supported assisted suicide for the dying: Survey flawed through inadequate definition of “religious” and “terminally ill”.
Those who “said that they followed a religion” were regarded as “religious followers” [1] and their opinions about “a change in the law to allow assisted suicide for people who are terminally ill” recorded in a survey which I consider deeply flawed because of inadequate definition of “religious” and, when Palliative Care Experts are not themselves unanimous [2] in agreeing on who can be called “terminally ill”, how was the term explained to the “nearly 4500 adults” in the survey? [1]
CHURCH OF ENGLAND QUERIES SURVEY
While it was claimed that there was “strong support for a change in the law” from 72% of 1519 Anglicans in the survey organised by Linda Woodhead, Lancaster University Professor of Sociology of Religion, the Church of England spokesman, quite rightly in my view, said such an important subject “cannot be effectively conducted through the medium of online surveys” – a criticism that Professor Woodhead apparently dismissed on the grounds that she was better placed to define who an Anglican follower was than the Church herself.
ETHICS, NOT SOCIOLOGY, IS CRUX OF THE MATTER
The second flaw in Professor Linda Woodhead’s survey is a lack of distinction between legality and ethics when it comes to questions of assisted suicide. To assume that “the Law” is all that needs consideration in this matter is grossly mistaken. I have previously pointed out in the BMJ how two brilliant British Professors of Genetics, both FRS, treated the ethical principle surrounding a genetic defect I had described in the BMJ, in two entirely different ways in their well known textbooks of Genetics. One FRS used my story to underline the importance of Ethics in Clinical Medicine. The other FRS, also publishing details of my story, did not even mention the word “ethics” in his Genetics book. Assisted suicide is bigger than sociology of religion. Any survey done without highlighting the ethical dimension, regardless of the brilliance and reputation of the organisers of the survey, is deeply flawed.
RELIGION IS BROADER THAN CHURCH, MOSQUE, SYNAGOGUE
Third Flaw: Professor Linda Woodhead needs no reminding that there are atheists who are deeply religious. Some of my brilliant atheistic friends and acquaintances worship Scientific Humanism. If they are also labelled as religious (as they need to be) then does the thrust of the survey with the message it seems to convey not become meaningless?
FACE-TO-FACE SURVEYS PREFERABLE IN CONTROVERSIAL MATTERS
In controversial matters it is always good policy to demand face to face interviews where the interviewer can be asked questions such as: (i) “What is your definition of ‘religion’?” (ii) “Have some terminally ill patients not been known to improve unexpectedly and gone home?” (iii) “Why was the present law on assisted suicide acceptable a decade ago, but not now?” (iv) “If Assisted Suicide is made legal in the UK does that make it ethical?” (v) “Does Hitler’s Nazi Germany have nothing to teach Great Britain?”
Felix I D Konotey-Ahulu MD(Lond) FRCP DTMH [felix@konotey-ahulu.com]
Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathies, 9 Harley Street Ltd, Phoenix Hosp[ital Group, London W1G 9AL
Competing interests: I am Christian but I do not refer to myself as “religious”.
1 Kmietowicz Zosia. Most religious followers support assisted suicide for the dying. BMJ 201`3; 346: f2855 (11 May, page 3) www.bmj.com/content/346/bmj.f2855?sso
2 Konotey-Ahulu FID. Liverpool care pathway BMJ and Channel Four News: Majority expert choice does not mean best choice.www.bmj.com/content/346/bmj.f1303/rr/634971 March 8 Rapid Response to “Nine out of 10 palliative care experts would choose Liverpool care pathway for themselves” Krishna Chinthapalli in BMJ 2013; 346: 1103 (March 2, pages 2-3)
2012
311 Konotey-Ahulu FID. Epistaxis from sickle cell disease must not be forgottenwww.bmj.com/content/344/bmj.e1097/rr/576087 BMJ Rapid Response 28 March 2012
312Konotey-Ahulu FID. World Sickle Cell Day 19th June 2012 www.sicklecell.md/blog/?p=132Featuring (i) The Inheritance of Sickle Cell Disease (ii) The Person with Sickle Cell Disease (iii) The Teenagwer with Sickle Cell Disease (iv) The Adult with Sickle Cell Disease.
313 Konotey-Ahulu FID. Further Communication on “Sickle Cell Trait Misinformation and Disinformation” and Sickle Cell Terminology: Disease or Disorder? www.sicklecell.md/blog/?p=127April 16 2012
314Konotey-Ahulu FID. Should clinicians edit Wikipedia to engage a wider world web? At least two examples of inaccuracy dictate caution www.bmj.com/content/345/bmj.e4275/rr/598116 BMJ 14 August 2012 Rapid Response
315Konotey-Ahulu FID. Management of an acute painful sickle cell episode in hospital: NICE guidance is frightening1 Sept 7 2012 www.bmj.com/content/344/bmj.e4063/rr/599158 [42 references]
316Konotey-Ahulu FID. Almost a quarter of Royal College Fellows say their hospitals cannot deliver continuity care. And they boast of something called National Institute of Clinical Excellence? www.bmj.com/content/345/bmj.e4942/rr/601191 September 7 2012 BMJ Rapid Response
317Konotey-Ahulu FID. There is no evidence that I was born on a Saturday. PRIVATE THOUGHTS – Postgraduate Medical Journal of Ghana 2012 (September); Volume 1, Number 1, pp 32-33 [Pointing out that the increasing use of “There is no evidence that ..” in scientific debate is itself unscientific]
318Konotey-Ahulu FID Bring back good quality paper in the print BMJwww.bmj.com/content/345/bmj.e6396/rr/610395 BMJ Rapid Response 23 October 2012
2013
319Konotey-Ahulu FID. Diagnosis and management of pulmonary embolism.www.bmj.com/content/346/bmj.f767/rr/633072 BMJ Rapid Response 26 Feb 2013
320Konotey-Ahulu FID. Liverpool care pathway BMJ and Channel Four News: Majority expert choice does not mean best choice. March 8 2013 www.bmj.com/content/346/bmj.f1303/rr/634971BMJ Rapid Response to “Nine out of 10 palliative care experts would choose Liverpool care pathway for themselves” Krishna Chinthapalli BMJ 2013; 346: 1103 (March 2, pages 2-3)
321 Konotey-Ahulu FID. Christianity and Africa. New African 2013 March, page 4http://bit.ly/Z0eb1K
322Konotey-Ahulu FID. Importance of history in the diagnosis of pulmonary embolismwww.bmj.com/content/346/bmj.f1692 March 19
BMJ 2013; 366: F1692
For World Sickle Cell Day 19th June 2012 it is worth reminding ourselves of the 4 consecutive articles I wrote for New African (London) in Jan/March/June/September 2001.
Click to view PDF Document



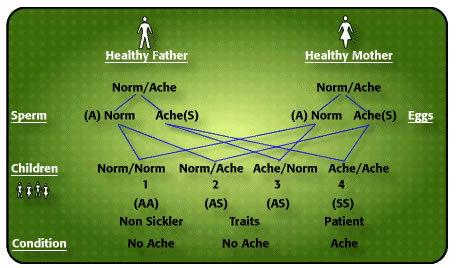

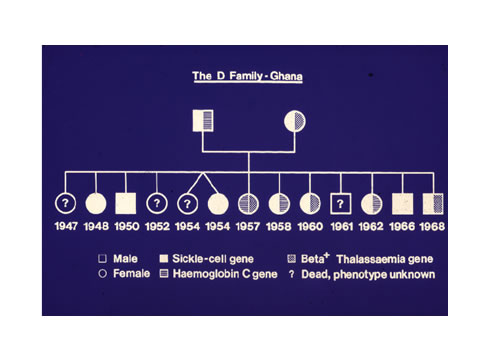





{kind=link}
{kind=link}
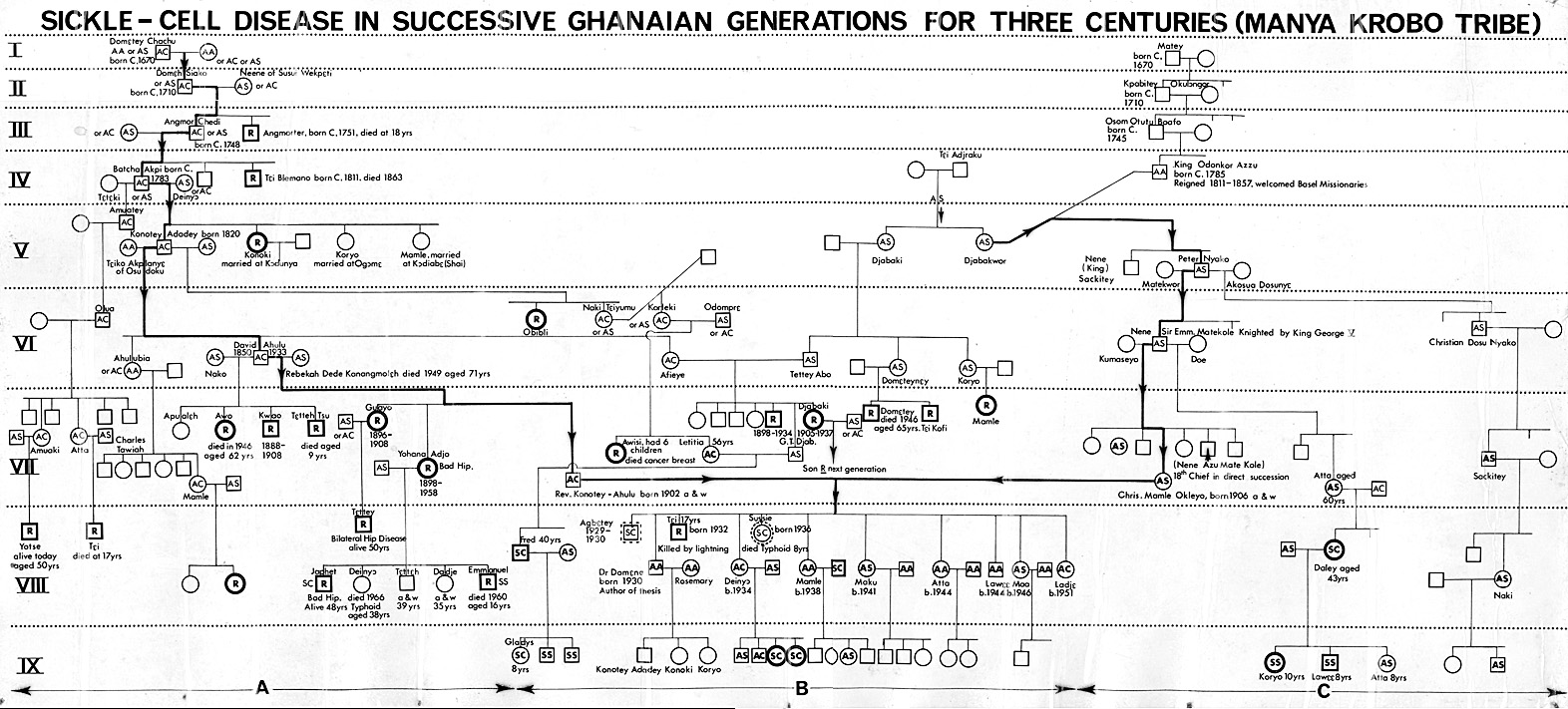{kind=link}
{kind=link}