Sickle Cell Trait Misinformation and Disinformation
F I D Konotey-Ahulu MB BS MD(London) FRCP FGCP FWACP FTWAS DTMH
Just google or wikipediate “sickle cell trait” and you get a mixture of truth and error which latter has led to (i) selective abortion (ii) insurance injustice (iii) social humiliation (iv) sports ban (v) employment exclusion (vi) clinical mismanagement and (vii) coroners’ perversion of truth. When misinformation turns “sickle cell trait” into “sickle cell disease” the consequences can be dire. Disinformation is deliberate misinformation. When terms like “congenital-abnormality” and “genetic-defect” mean different things to different people, confusion reigns. Wrong ethics plus wrong definitions of congenital or genetic defect make me revisit “abortion for sickle cells” and the Sickle Cell Trait Controversy.
WHO DEFINES “CONGENITAL ABNORMALITY” THAT NEEDS ABORTION?
“When fetal congenital abnormalities are present abortion is a valid therapeutic medical procedure” [1] was Dr Saripanidis’ response to Helen Watt who found it “heartening that so many medical students show at least some unwillingness to carry out abortion, a procedure which lethally affects the health of the fetal child (congenitally abnormal or otherwise”) [2]. Saripanidis’ “congenital abnormality” may be different from others’. Watt was responding to Zosia Kmietowicz who had reported “a fifth of medical students object to abortion for congenital abnormality” [3].
ETHICS DIVIDES EQUALLY BRILLIANT EXPERTS
I once described my Mendellian Dominant extra digits as being of no consequence, but which for a tribe east of us a child born with them was, to our disgust, promptly drowned [4]. Two Fellows of the Royal Society read my likening British doctors who advised prenatal diagnosis and selective abortion for sickle cell disease to “scientific tribesmen” behaving in a way that we Krobo people found reprehensible. Publishing my case in their Genetics books Sir David Weatherall mentioned the ethical implications in a chapter on ETHICS [5]. The other professor did not bother to mention Ethics in relation to Genetics [6]. My own position is well known. [4, 7-12] Pregnancies have been terminated for cleft palate. Extra digits are liable to be prenatally detected and fetus aborted. Making sickle cell trait a disease exposes it to an abortion programme because the received wisdom in the UK is prenatal diagnosis with advice for selective abortion.
SICKLE CELL TRAIT CONFUSED WITH SICKLE CELL DISEASE
Sickle Cell Trait (1 normal ß-globin gene ‘A’ plus 1 ß-globin variant gene ‘S’) is being confused with Sickle Cell Disease (2 ß-globin variant genes at least 1 of which is the sickle cell gene ‘S’). Look at these statistics:
(a) Among over 170 million Nigerians more than 35 million have Sickle Cell Trait ‘AS’.
(b) Ghana’s population of 25 million has 5 million sickle cell traits ‘AS’
(c) Of 6000 medically qualified West African doctors (Nigerian 4,500 and Ghanaian 1,500) working in the USA 1,500 have Sickle Cell Trait (‘AS’)
(d) One in 5 of Nigerian, Ugandan, and Ghanaian doctors in the UK are sickle cell trait.
(e) Korle Bu Teaching Hospital in Accra sees over 100,000 patients annually of whom 20,000 are Sickle Cell Trait (‘AS’)
(f) One in 5 diabetics, 1 in 5 hypertensives, 1 in 5 liver failures, 1 in 5 kidney failures, 1 in 5 suddenly-dropped-down-deads, and 1 in 5 stammerers have the sickle cell trait “AS”, because 1 in 5 of the southern Ghanaian healthy population is sickle cell trait “AS”.
Against this background we have:
(a) Elliott Vichinsky: “Renal medullary carcinoma is a rare and aggressive tumor that is seen almost exclusively in young patients with sickle cell trait” [13].
(b) Charis Kepron, Gino Somers, Michael Pollanen: “Sickle Cell Trait Mimicking Multiple Inflicted Injuries in a 5-Year-Old Boy” [14]
(c) Nigel Key, Vimal Derebail: “During exercise, Sickle Cell Trait appears to be a risk factor for sudden death and/rhabdomyolysis, particularly when the exercise is intense, and is performed at high altitude …” [15]
(d) Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong, Y: “Complications associated with sickle cell trait: a brief narrative review”. [16]
So 100 million African sickle cell traits, millions in southern Turkey (1 in 5 Eti-Turks are ‘AS’), Mediterranean people with millions of ‘AS’ Trait [17-20], plus millions in India’s Madhya Pradesh and Orissa with 43% ‘AS’ Trait [21] are in some danger? Insurance injustice apart, the next step would be to advise aborting any fetus with Hb ‘S’ in it.
“UP-TO-DATE” AND “NOVEL” INSIGHTS ARE OUT OF DATE
Forty years ago exactly the then BMJ Editor Dr Martin Ware helped to retract a sickle cell trait inaccurate publication that caused furore [22-29]. No author has mentioned the 10 Addae’s Criteria required to be fulfilled before someone attributes symptomatology to sickle cell trait [24]. Witkowska et al published that sickle cell trait and sickle cell disease co-existed in the same person [30]. Elliott Vichinsky [13] failed to pick up the genetic heresy of 4 ß-globin genes in one person. I had warned that Witkowska’s patient did not have sickle cell trait but ‘Sickle Cell Haemoglobin Quebec-Chori’ disease [31] which “made it vital for clinicians to probe further any case of sickle cell trait where the symptoms suggest sickle cell disease” [31]. To provide excuse to include the sickle cell trait phenotype in an abortion programme is frightening.
CLINICAL EXPERIENCE MUST SUPERINTEND HAEMATOLOGICAL EXPERIENCE
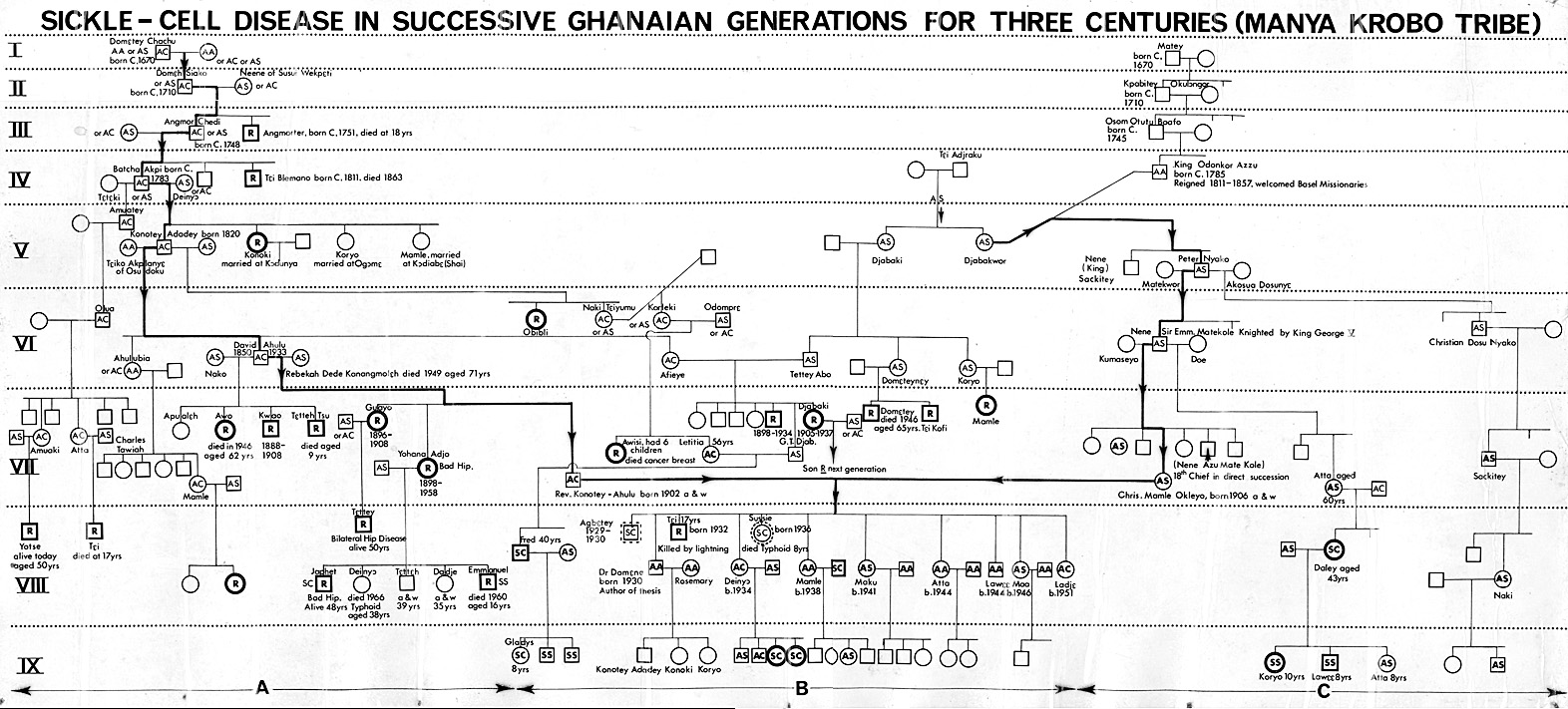
My views here are not those of a novice. Professor Helen Ranney once said: “There is no single clinical experience in the United States comparable to that of Dr Konotey-Ahulu” [32]. Our family named sickle cell disease patients from 1670 Anno Domini http://www.konotey-ahulu.com/images/generation.jpg correctly identifying phenotypes Chwechweechwe/Hemkom (Sickle cell Disease) “SC” as “pi-gbagblaa”, and “SS” as “gbagblaa” [33 34 38]. Directing the largest Sickle Cell Disease Clinic in the world [34], I could guess the 4 sickle cell disease phenotypes correctly (‘SS’, ‘SC’, SF’, SßThal’) without haemoglobin electrophoresis [34 35]. That was how Haemoglobin Korle-Bu and Haemoglobin Osu-Christiansborg were discovered [36, 37]. We even knew that the commonest cause of severe sickle cell crisis was malaria [34 39], yet erroneous statements like “Sickle Cell Disease protects against malaria” persist.
HISTORY BEHIND CONFUSION WITH SICKLE CELL TRAIT DEFINITION
When Itano discovered Haemoglobin C in 1951 [40], the riddle was solved of seeing sickle cell disease symptomatology in someone who was wrongly routinely referred to as “Sickle Cell Trait” (1 ß-globin gene variant ‘S’ plus 1 normal ß-globin ‘A’) because the blood of one of the parents of the “SC disease phenotype” (2 ß-globin gene variants) did not sickle. Moreover when the ‘AS’ pattern represents 2 abnormal genes as in Sickle Cell ß-plus Thalassaemia, or as in Sickle Cell Haemoglobin Quebec-Chori disease, clinical experience more than haematological experience is what is required to prevent wrong interpretations. [Read that again, and again, please]
Furthermore, the sickle cell colour test does not distinguish between Sickle Cell Trait ‘AS’ and Sickle Cell Hb C disease ‘SC’ [41], and has thus wrongly attributed death to Sickle Cell Trait. I was once guilty of rushing to publication only to be corrected by Dr E J Watson-Williams who advised Family Studies [34, p. 364]. I then discovered to my shame [42] that the man who had died in London during a minor eye operation was “SC phenotype”, not sickle cell trait “AS” phenotype.
IMPLICATIONS FOR SICKLE CELL TRAIT REDEFINED AS SICKLE CELL DISEASE
ABORTION ADVICE is one consequence. INSURANCE PUNISHMENT is another. I was given 4 body guards in Philadelphia for my Keynote Address at The Martin Luther King Jr Foundation Award Ceremony. The title was “Difference between Sickle Cell Trait and Sickle Cell Disease” [43]. Insurance companies wanted the difference blurred. Approached by Professor Bowman in the USA about the Sickle Cell Trait [44] they admitted to loading the trait’s premium. [44] “It is my understanding” says Bowman “that insurance companies test only Blacks for the sickle cell trait” [45]. Did these authors (13-16) exclude Hb Quebec-Chori masquerading as normal Hb ‘A’? Was Hb ‘S’ quantified in all their publications? How many patients had G6PD quantified as 1 in 5 Ghanaian males and 1 in 16 females have total G6PD Deficiency [46 47 48] which affects the kidney adversely [49 50 51]? How many Family Studies were done?
IN SPORT ARE ALL NATIONALITIES PHENOTYPED “FOR THEIR OWN SAFETY”?
Are white skinned Americans included in sickle cell screening before sports? Nobel Laureate Professor James Watson’s great grand parent was an African. [52 53] As 1 in 3 West Africans had a ß-globin gene variant ‘S’ or ‘C’ [10] could Watson have either? Regarding advice given [22] to screen “Negro travellers” at airports for sickle cells for their own safety Dr Djabanor asks regarding White sickle cell traits: “How do we identify them from their external features to thrust upon them the benefits of this advice?” [23]
HAEMOGLOBIN ‘S’ IN TRUE SICKLE CELL TRAIT IS BETWEEN 20% AND 39.7%
Of 82 consecutive sickle cell traits that emerged out of over 400 consecutive West Africans I saw in London that Professor Hermann Lehmann phenotyped for me the 3 known bands of Hb ‘S’ percentages were clearly demonstrated, with the highest percentage less than 40%, and the lowest haemoglobin ‘S’ value was as little as 20% [54]. Are these the levels of Hb S that we are told millions around the world stand in danger from? Writing to The London Times exactly 40 years ago Lehmann confirmed that sickle cell traits competed in the Olympic Games in Mexico City at 7000 ft above sea level and no sudden deaths occurred. Chicago University’s Professor James Bowman said : “Persons with sickle cell trait will no longer be able to become ill or even die lest they find themselves subject of case report” [55], and a Black man beaten to death by police was claimed by the Coroner to have died from sickle cell trait. Professors Simon Dyson and Gwyneth Bosswell recount similar experiences [56]. Social humiliation resulted with suggestion [22] that “Negro travellers” be tested at airports for sickle cells before flight “for their own safety”. Acute appendicitis was misdiagnosed as abdominal sickle cell crisis because sickle cell test was positive, and employment was refused on the grounds of “sickle cell trait”. I found 15 flaws [57] in the article on the 5-year old boy who died and was found with “multiple inflicted injuries” and aspiration pneumonia, only to be published as “Sickle cell trait mimicking multiple inflicted injuries” [14]. My Sickle Cell Trait chapter [34 pp 349-371] deals comprehensively with similar anomalies.
BALANCED POLYMORPHISM AND AFRICAN ANTHROPOGENETICS
We know that the true ‘AS’ [NORMACHE] can be tougher than phenotype ‘AA’ [NORMNORM] because in the toddler age group we find no cerebral malaria child with sickle cell trait [58], which fact is the basis of the Balanced Polymorphism phenomenon [34 pp. 91-108], about which there is so much ignorance [59]. To assess the Hardy Weinberg Equation of gene frequencies without reference to the genetic index ‘MPSI’ that I once invented, and to polygamy [60 61 62], is to ignore African realities.
TRANSPARENCY AND ABSENCE OF PREJUDICE REQUIRED
Why all of a sudden such concern that “for their own safety” sickle cell traits should not be allowed to run competitively? And has there been an attempt to find out why Black families are increasingly refusing prenatal screening in the UK? [63] Is this reluctance related to the rumour that a Teaching Hospital screened a West African mother who refused abortion when told baby would be ‘SS’ but when born was found to be ‘AA’, and “Laboratory Error” was claimed as reason for the misinformation 7 months previously? [63] Medical Practice is known to have been marred in time past by racial prejudice [64 65 66 67], so one needs to ask what proportion of the medical students “object to abortion for congenital abnormality” [1 2 3] not just for ethical reasons but also because they are from ethnic minority groups that harbour suspicions?
Competing interests: I not only carry a congenital abnormality, Mendellian dominant extra manual digits, but I am also one of 11 children of parents both of whom were Traits for beta-globin variant genes [NORMACHE], resulting in 3 siblings born with Sickle Cell Disease [ACHEACHE], 4 with Trait [NORMACHE] and 4 with “AA” [NORMNORM] – see www.konotey-ahulu.com/images/generation.jpg
Felix I D Konotey-Ahulu MD(Lond) FRCP DTMH Kwegyir Aggrey Distinguished Professor of Human Genetics, University of Cape Coast, Ghana and Consultant Physician Genetic Counsellor in Sickle Cell and Other Haemoglobinopathies, 9 Harley Street, Phoenix Hospital Group, London W1G 9AL [E-mail: felix@konotey-ahulu.com
Website: http://www.sicklecell.md]
1 Saripanidis Stravos. Abortion is a valid therapeutic medical procedure. BMJ Rapid Response 30 October 2011. Re: BMJ 2011; 343.doi: 10.1136/bmj.d4717.1
2 Watt Helen. Doctors should not refer for harmful elective procedures. Rapid Response 26 July 2011. Re: BMJ 2011; 343.doi: 10.1136/bmj.d4717.1
3 Kmietowicz Zosia. A fifth of medical students object to abortion for congenital abnormality. 22 July 2011. BMJ 2011; 343.doi: 10.1136/bmj.d4717.1
4 Konotey-Ahulu FID. Ethical issues in prenatal diagnosis. BMJ Clin Res Ed 1984; 289(6438)185 http://www.bmj.com/cgi/reprint/289/6438/185-a.pdf July 21 doi:10.1136/bmj.289.6438.185-a
5 Weatherall D J. Ethical issues and related problems arising from the application of the new genetics to clinical practice. Chapter 12 In The New Genetics and Clinical Practice. Oxford University Press (Third Edition) Oxford 1991, pages 347-48.
6 Jones Steve. The Language of Genes; Biology, History, and Evolutionary Future. Flamingo 1993, London, page 285.
7 Konotey-Ahulu FID. Ante-natal diagnosis of haemoglobinopathies. Lancet 1977; 1: 597-598.
8 Konotey-Ahulu FID. Ethics of amniocentesis and selective abortion for sickle cell disease. Lancet 1982; 1(8262): 38-39. January 2.
9 Konotey-Ahulu FID. Missing the wood for one genetic tree? The First International Symposium on the Role of Recombinant DNA in Genetics – Proceedings – Chania, Crete, Greece, May 13-16 1985. Eds Loukopoulos D, Teplitz RL; Athens, P. Paschalidis 1986, pages 105-116.
10 Ringelhann B, Konotey-Ahulu FID. Hemoglobinopathies and thalassemias in Mediterranean areas and in West Africa: Historical and other perspectives 1910 to 1997 – A Century Review. Atti dell’Accademia dell Science di Ferrara 1998; 74: 267-307 Milan
11 Konotey-Ahulu FID. Antenatal screening for sickle cell disease and beta-thalassaemia. http://www.bmj.com/content/341/bmj.c5132/reply#bmj_el_242914 BMJ Rapid Response Oct 12 2010
12 Konotey-Ahulu FID. Antenatal sickle cell disease/haemoglobinopathy screening. BMJ Rapid Response http://www.bmj.com/content/341/bmj.c5243/reply#bmj_el_243447 October 25 2010
13 Vichinsky Elliott P. Sickle cell trait. Literature Review UpToDate [Accessed 18 Feb 2011] http://www.uptodate.com/contents/sickle-cell-trait?view=print
14 Kepron Charis, Somers Gino R, Pollamen Michael S. Sickle Cell Trait Mimicking Multiple Inflicted Injuries in a 5-Year-Old Boy. Journal of Forensic Sciences Volume 54, No.5, pp 1141 t0 1145 September 2009.
15 Key Nigel S, Derebail Vimal K. Sickle Cell Trait: Novel Clinical Significance. Hematology 2010: 418-422.
16 Tsaras, G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong, Y. Complications associated with sickle cell trait: a brief narrative review. American Journal of Medicine 2009; 122(6): 507-512.
17 Aksoy M. Sickle cell trait in Southern Turkey. Lancet 1955; 1: 589-90.
18 Atlay C, et al. Haemoglobin S and some other hemoglobinopathies in Eti-Turks. Human Heredity 1978; 28: 56-61.
19 Choremis C et al. Sickle cell anaemia in Greece. Lancet 1951; 1: 1147.
20 Choremis et al. Blood groups of a Greek community with a high sickling frequency. Lancet 1957; 2: 1333-34.
21 Roy DN, Chaudhuri RSK. Sickle cell trait in the tribal population in Madhya Pradesh and Orissa (India). Journal of Indian Medical Association, 1967; 49: 107-112.
22 Green RL, Huntsman RG, Serjeant GR. Sickle cell trait and altitude. Br Med J 1971; 4: 593-595.
23 Djabanor F F T. Sickle cell trait and altitude. Brit Med J 1972; 1: 113
24 Addae R O. Sickle cell trait and altitude. BMJ 1972; 1: 53. [10 criteria required to satisfy clinicians in regions where 1 in 5 people have the sickle cell trait that symptoms are due to the trait]
25 Konotey-Ahulu FID. Sickle cell trait and altitude. Brit Med J 1972; 1: 177-178.
26 Lehmann Hermann. Sickle cell and flying. The Times (London), 4th January 1972.
27 Konotey-Ahulu FID. An international sickle cell crisis. [Editorial] Ghana Medical J; 1972; 11: 4-8
28 Konotey-Ahulu FID. Sickle cell trait and altitude. BMJ 1972; 2: 231-32 April 22
29 Green RL, Huntsman RG, Serjeant GR. Sickle and altitude. Brit Med J. 1972; 2: 294
30 Witkowska HE, Lubin BH, Beuzard Y et al. Sickle cell disease in a patient with sickle cell trait and compound heterozygosity for haemoglobin S and haemoglobin Quebec-Chori. New England Journal of Medicine 1991; 325: 1150-1154. [Note that the title of this article is incorrect: No human being can be said to have both Sickle cell trait and Sickle Cell Disease. The ‘AS’ pattern is sickle cell trait pattern, but this ‘A’ is not a true ‘A’ but the new haemoglobin called Quebec-Chori, producing a disease phenotype, not trait.]
31 Konotey-Ahulu FID. Beware of symptomatic sickle cell traits. Lancet 1992; Feb. 29, p 565. [Re: The enigma of Haemoglobin Quebec-Chori]
32 Ranney Helen M. Summary of Symposium. In Sickle Cell Disease, Editors H Abramson, J F Bertles, Doris Wethers [C Mosby Co., St Louis] 1973, p 320.
33 Konotey-Ahulu FID. Sickle cell disease in successive Ghanaian generations for three centuries. Chapter 2 pp 6-20 in The Sickle Cell Disease Patient: Natural History from a Clinico-epidemiological study of the first 1550 patients of Korle Bu Hospital Sickle Cell Clinic. Macmillan Press Ltd, London 1991 & 1992 and T-A’D Co Ltd Watford 1996.
34 Konotey-Ahulu FID. The Sickle Cell Disease Patient: Natural History from a Clinico-epidemiological study of the first 1550 patients of Korle Bu Hospital Sickle Cell Clinic. Macmillan Press Ltd, London 1991 & 1992 and T-A’D Co Ltd Watford 1996
35 Konotey-Ahulu FID. Patterns of clinical haemoglobinopathy. E Afri Med J 1969 Mar; 46(3): 149-156. PMID: 5800410 [PubMed – indexed for MEDLINE]
36 . Konotey-Ahulu FID, Gallo E, Lehmann H, Ringelhann B. Haemoglobin Korle Bu (alpha2 beta2 73 Aspartic Acid –> Asparagine), showing one of the two amino acid substitutions of Haemoglobin C Harlem. J Med Genet 1968 June; 5(2): 107-111. & http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1468514 An example of intra-genic cross-over [Also see G-Accra http://lib.bioinfo.pl/pmid:5722880 ]
37 Konotey-Ahulu FID, Kinderlerer, JL Lehmann H and Ringelhann B. Haemoglobin Osu-Christiansborg. A new chain variant of Haemoglobin A (beta 52 D3 Aspartic Acid –> Asparagine) in combination with Haemoglobin S. Journal of Med Genet 1971 Sep; 8(3): 302-305. http://pubmedcentral.nih.gov/picrender.fcgi?artid=146917&blobtype=pdf or http://lib.bioinfo.pl/pmid:5097135
38 Konotey-Ahulu FID. The Sickle-cell Diseases: Clinical manifestations including the Sickle Crisis http://archinte.ama.assn.org/cgi/reprint/133/4/611-pdf . Arch Intern Med 1974; 133(4): 611-619.
39 Konotey-Ahulu FID. Malaria and sickle-cell disease BMJ 1971 June; 2(5763): 710-711 doi:10.1136/bmj.2/5763.710-d http://www.bmj.com/cgi/reprint/2/5763/710-d.pdf
40 Itano HA. A third abnormal haemoglobin associated with hereditary hemolytic anemia. Proc Nat Acad Sci (Washington) 1951; 37: 775-84
41 Konotey-Ahulu FID. Detecting sickle cell haemoglobin. Brit Med J 1972; 4: 239
42 Konotey-Ahulu FID. Blushing in the black skin. (Invited Editorial) Journal of Cosmetic Dermatology April 2003; 2 (2), 59-60 [PMID: 17156057 PubMed]
43 Konotey-Ahulu FID. Four bodyguards and the perils of unmasking scientific truths. BMJ 2007; 335: 210-211. http://www.bmj.com/cgi/content/full/335/7612/210 (July 28)
doi:10.1136/bmj.39268.553021.47 Print http://www.bmj.com/cgi/reprint/335/7612/210.pdf
[Day & Date: Wednesday 31st May 1972 – Philadelphia, Dr Martin Luther King Jr Foundation Award Ceremony for Outstanding Contributions in Sickle Cell Disease: Banquet – My Keynote Address was on ‘Difference between Sickle Cell Trait and Sickle Cell Disease’. Those also honoured present on the platform with me included Nobel Prize Winners Linus Pauling and Max Perutz, then Hermann Lehmann, Roland Scott, J V Neel, Bella Ringelhann, A C Allison, James Bowman, Helen Ranney, Charles Whitten, Samuel Charache, L Diggs, L Conley, Sam Charache & Graham Serjeant].
44 Konotey-Ahulu FID. Insurance and genetic testing. Lancet 1993, 341: 833. March 27
45 Bowman James. Ethical, legal, and humanistic implications of sickle cell programs. INSERM 1975; 44: 353-378.
46 Ringelhann B, Dodu SRA, Konotey-Ahulu FID and Lehmann H. A survey for haemoglobin variants, thalassaemia and Glucose-6-Phosphate Dehydrogenase Deficiency in Northern Ghana. Ghana Med J 1968; 7: 120-124.
47 Konotey-Ahulu FID. Hereditary qualitative and quantitative erythrocyte defects in Ghana: An historical and geographical survey. Ghana Med J 1968; 7: 118-119 (Editorial 35 references). [First time Chwechweechwe and other tribal names were identified as Sickle Cell Disease]
48 Owusu SK. Glucose 6 Phosphate dehydrogenase (G6PD) deficiency in the causation of disease in Ghana. Ghana Med J. 1974; 13: 168-70
49 Owusu SK. Absence of glucose 6 phosphate dehydrogenase in red cells of an African. Brit Med J 1972; 4: 25-26
50 Owusu SK, Addy JH, Foli AK, Janosi M, Konotey-Ahulu FID and Larbi EB. Acute reversible renal failure associated with glucose-6-phosphate dehydrogenase deficiency. Lancet 1972 June 10; 1(7763): 1255-1257.
51 Adu D et al. Acute renal failure and typhoid fever in Ghana. Ghana Med J 1975; 14: 172-74.
52 Verkaik Robert. Scientist who sparked racism has black genes. The Independent, London. 10 December 2007. [Re: DNA Nobel Laureate Professor James Watson]
53 Konotey-Ahulu FID. There is but one human race. New African, London. Dec 2009, No. 490, page 4.[Re: James Watson who with Francis Crick won Nobel Prize on DNA]
54 Konotey-Ahulu FID. Alpha-Thalassaemia nomenclature and abnormal haemoglobins Lancet 1984; 1: 1024-25 May 5 [“Of 82 consecutive sickle cell traits seen in London in 24 months 36 (44%) had just one quarter of the total haemoglobin as sickle haemoglobin (mean 25%, range 20-28%) ..The three known peaks of haemoglobin S proportion in the West African sickle cell sickle cell trait are around 25%, around 30& and around 38%”)
55 Bowman JE, Bernstein S. Caution about preliminary reports. Pediatrics 1977; 59: 639-40.
56 Dyson Simon, Bosswell Gwyneth. Sickle Cell and Deaths in Custody. Whiting and Birch, London: June 2009; 230 pages “The misuse of Sickle Cell Trait to explain away sudden deaths”.
57 Konotey-Ahulu FID. Blaming sudden death on Sickle Cell Trait? Flaws in article of Charis Kepron, Gino Somers and Michael Pollanen Exposed. September 4 2011. . www.sicklecell.md/blog/?p=105 or www.konotey-ahulu.com/blog/?p=105
58 Commey JOO. Absence of sickle cell trait in 30 consecutive cases of cerebral malaria in Ghana when 6 expected. [Observations in 1986 see Reference 33, page 95] See also Konotey-Ahulu FID. Balanced polymorphism and other factors relating to hereditary qualitative and quantitative erythrocyte defects. Chapter 8, pp 91-108 in Reference 34, and also Chapter 30 Percentage values of haemoglobins S, F, A2 C, A, in various sickle cell phenotypes, and a consideration of the sickle cell trait pp 349-370]..
59 Konotey-Ahulu FID. Malaria and sickle cell: “Protection?” Or “No Protection?” – Confusion reigns. http://www.bmj.com/cgi/eletters/337/oct01_3/a1875#203067 BMJ Rapid response 13 October 2008
60 Konotey-Ahulu FID. Male procreative superiority index (MPSI): The missing co-efficient in African anthropogenetics. BMJ 1980; 281(6256): 1700-1702 http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=1715685&blobtype=pdf doi:10.1136/bmj.281.6256.1700 December 20 – 27 1980 http://www.bmj.com/cgi/reprint/281/6256/1700.pdf
61 Konotey-Ahulu FID. The Male Procreative Superiority Index (MPSI): its relevance to genetical counseling. In FIFTY YEARS OF HUMAN GENETICS A Festschrift and liber amicorum to celebrate the life and work of GEORGE ROBERT FRASER Edited by Oliver Mayo and Carolyn Leach. Wakefield Press 2007 (www.wakefieldpress.com.au) 1 The Parade West, Kent Town, South Australia 5067)
62 Bonney GE and Konotey Ahulu FID. Polygamy and genetic equilibrium. Nature 1977; 265: 46-47 (January 6 1977). doi:10.1038/265046a0
http://www.nature.com/nature/journal/v265/n5589/abs/265046a0.html
63 Konotey-Ahulu FID. Konotey-Ahulu FID. Refusing to provide a prenatal test for refusing later termination of pregnancy can it ever be ethical? BMJ Rapid Response November 20 2006 http://www.bmj.com/cgi/eletters/333/7577/1066#149662
64 Muller-Hill Benno. Murderous Science. Elimination by Scientific Selection of Jews, Gypsies, and Others – Germany 1933-1945. [Translated from German by G R Fraser] Oxford, Oxford University Press, 1988.
65 Clinton President WJ. Apology on behalf of the American Government to 8 survivors of the Tuskegee Syphillis experiment victims. World-wide radio and television PBS News Hour Newsreel Announcement (Jim Lehrer and Charlayne Hunter-Gault) May 16 1997
http://www.pbs.org/newshour/bb/health/may97/Tuskegee_5-16.html
66 Tanne Janice Hopkins. President Obama apologizes to Guatemala over 1940s syphilis study. BMJ 341: doi:10.1136/bmj.c5494 October 4 2010
67 . Konotey-Ahulu FID. President Obama apologizes over Guatemala syphilis study: International co-operative research and practice in jeopardy. Rapid response BMJ 17 October 2010 http://www.bmj.com/content/341/bmj.c5494.full/reply#bmj_el_243183


{kind=link}
{kind=link}